


A deficiência seletiva de imunoglobulina A ( IgA IgA Represents 15-20% of the human serum immunoglobulins, mostly as the 4-chain polymer in humans or dimer in other mammals. Secretory iga is the main immunoglobulin in secretions. Immunoglobulins: Types and Functions) é o tipo mais MAIS Androgen Insensitivity Syndrome comum de imunodeficiência primária. A condição é uma hipogamaglobulinemia caracterizada por uma ausência ou níveis reduzidos de IgA IgA Represents 15-20% of the human serum immunoglobulins, mostly as the 4-chain polymer in humans or dimer in other mammals. Secretory iga is the main immunoglobulin in secretions. Immunoglobulins: Types and Functions. Este anticorpo reside principalmente nas membranas mucosas da boca, vias aéreas e trato digestivo. A causa exata é desconhecida. A doença geralmente é assintomática, embora alguns doentes possam apresentar infeções respiratórias e gastrointestinais recorrentes, bem como doenças autoimunes e malignas. O diagnóstico é realizado através da medição de níveis excecionalmente baixos de IgA IgA Represents 15-20% of the human serum immunoglobulins, mostly as the 4-chain polymer in humans or dimer in other mammals. Secretory iga is the main immunoglobulin in secretions. Immunoglobulins: Types and Functions no soro, na presença de níveis normais de IgG IgG The major immunoglobulin isotype class in normal human serum. There are several isotype subclasses of igg, for example, igg1, igg2a, and igg2b. Hypersensitivity Pneumonitis e IgM IgM A class of immunoglobulin bearing mu chains (immunoglobulin mu-chains). Igm can fix complement. The name comes from its high molecular weight and originally being called a macroglobulin. Immunoglobulins: Types and Functions.
Last updated: Dec 15, 2025
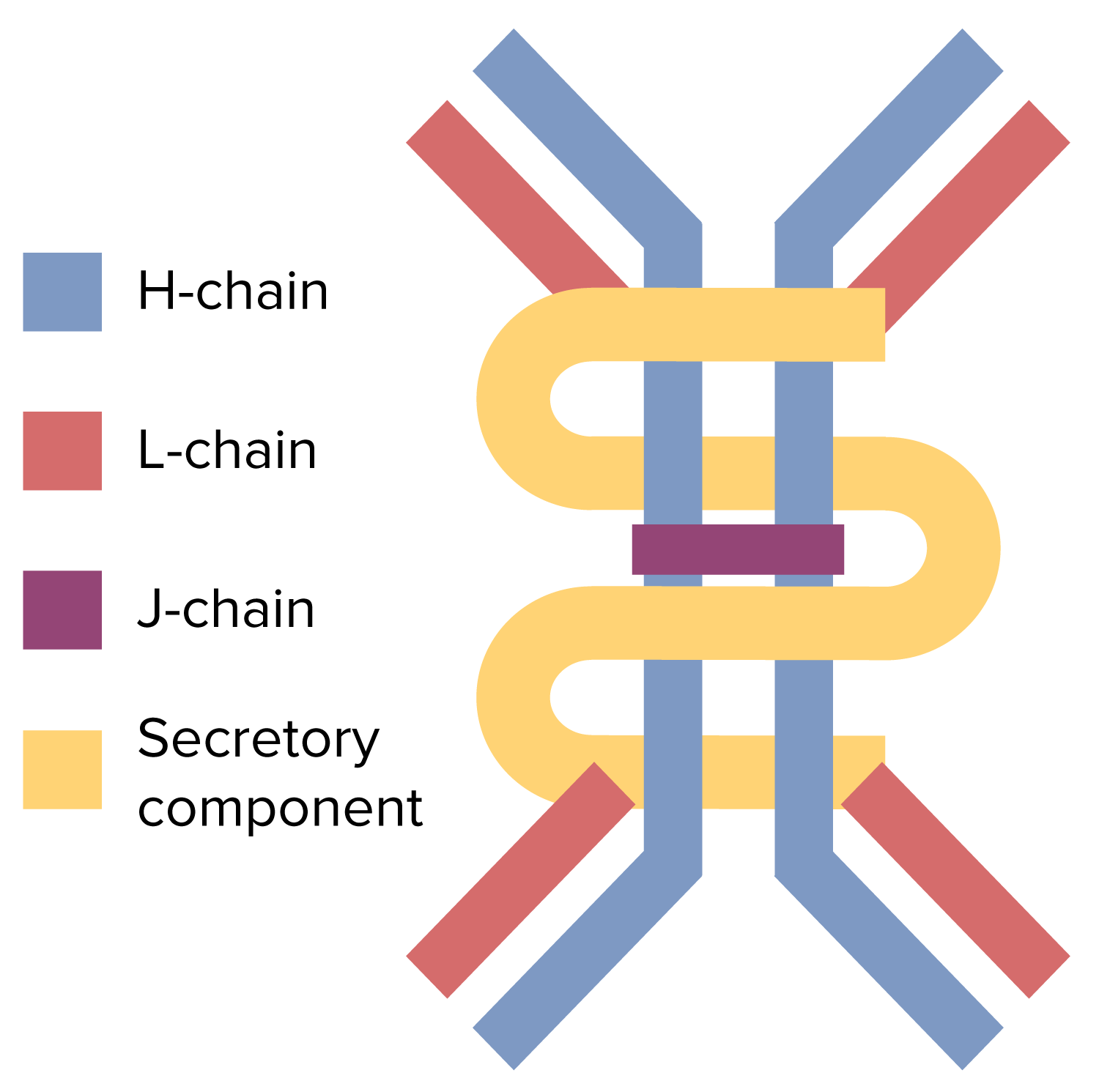
Estrutura dimérica do anticorpo IgA
Imagem por Lecturio.Aproximadamente 90% dos casos são assintomáticos.
Doentes sintomáticos podem apresentar uma mistura dos seguintes sintomas:
Diagnóstico
Tratamento
As condições a seguir são outras imunodeficiências congénitas de células B que servem como diagnósticos diferenciais na deficiência seletiva de IgA IgA Represents 15-20% of the human serum immunoglobulins, mostly as the 4-chain polymer in humans or dimer in other mammals. Secretory iga is the main immunoglobulin in secretions. Immunoglobulins: Types and Functions.
