El síndrome de Treacher Collins es una enfermedad genética rara con herencia autosómica dominante. El síndrome de Treacher Collins también se conoce como disostosis mandibulofacial o síndrome de Franceschetti y se caracteriza por importantes deformidades craneofaciales y pérdida de audición conductiva. El síndrome de Treacher Collins es una enfermedad estrictamente física y no afecta a la cognición ni a otras esferas del desarrollo. El diagnóstico se confirma con pruebas genéticas. El tratamiento de las vías respiratorias, la capacidad de alimentación y la pérdida de audición son las principales preocupaciones en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum los LOS Neisseria primeros años de vida. En EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum última instancia, es necesaria la cirugía reconstructiva facial. La esperanza de vida es normal.
Last updated: Dec 15, 2025
Este síndrome se caracteriza por una multitud de defectos y anomalías estructurales craneofaciales bilaterales y a menudo asimétricas.
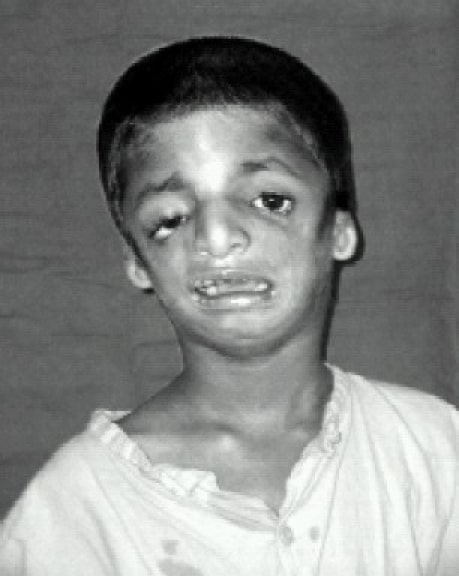
Paciente que muestra hipoplasia de los huesos faciales, fisuras palpebrales inclinadas hacia abajo y coloboma de los párpados inferiores con escasas pestañas inferiores
Imagen: “Front view” por Indian J Anaesth. Licencia: CC BY 2.0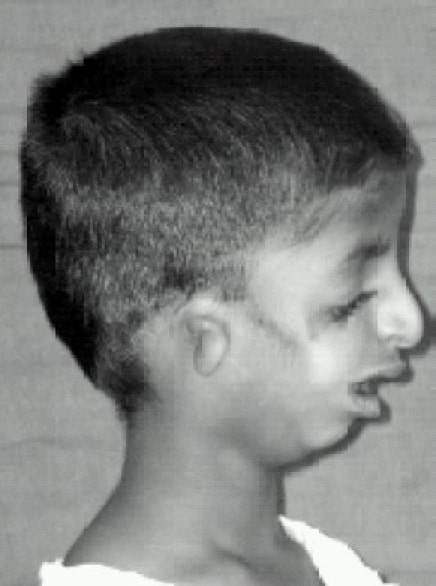
Vista lateral de la paciente que muestra la deformación de la oreja (microtia) y la retrognatia
Imagen: “Lateral view” por Indian J Anaesth. Licencia: CC BY 2.0Las malformaciones craneofaciales pueden dar lugar a un compromiso de las vías respiratorias y a dificultades de alimentación, por lo que el tratamiento de estos problemas es prioritario en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum los LOS Neisseria primeros años. Una vez resueltos los LOS Neisseria problemas que amenazan con la vida, la gestión de la pérdida de audición es imprescindible para el desarrollo del habla. La cirugía es necesaria para corregir las anomalías faciales.