


A síndrome de Treacher Collins é uma condição genética, rara, com hereditariedade autossómica dominante. A síndrome de Treacher Collins, também conhecida como disostose mandibulofacial ou síndrome de Franceschetti, é caracterizada por deformidades craniofaciais significativas e perda auditiva de condução. A síndrome de Treacher Collins é estritamente uma doença física e não afeta a cognição ou outras esferas do desenvolvimento. O diagnóstico é confirmado com testes Testes Gonadal Hormones genéticos. A abordagem da via aérea, a capacidade de alimentação e a perda auditiva são as principais preocupações nos primeiros anos de vida. Em última análise, é necessária a cirurgia reconstrutiva facial. A expectativa de vida é normal.
Last updated: Dec 15, 2025
Esta síndrome é caracterizada por uma multiplicidade de defeitos e alterações estruturais craniofaciais bilaterais e muitas vezes assimétricos.
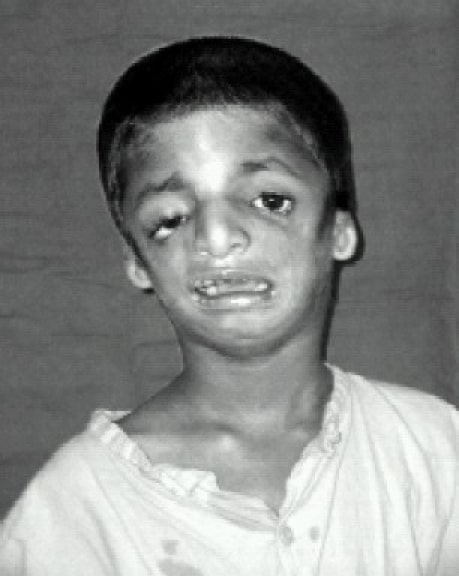
Doente a demonstrar hipoplasia dos ossos faciais, fissuras palpebrais inclinadas para baixo e coloboma das pálpebras inferiores com escasses dos cílios inferiores
Imagem : “Front view” por Indian J Anaesth. Licença: CC BY 2.0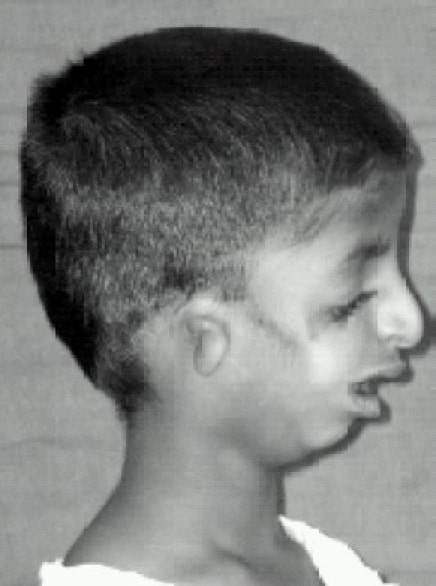
Visão lateral do doente a mostrar deformidade da orelha (microtia) e retrognatia
Imagem: “Lateral view” por Indian J Anaesth. Licença: CC BY 2.0As malformações craniofaciais podem resultar em comprometimento das vias aéreas e dificuldades de alimentação, tornando a abordagem destes problemas a prioridade nos primeiros anos. Uma vez resolvidos os problemas com risco de vida, a abordagem da perda auditiva é imperativa para o desenvolvimento da fala. A cirurgia é necessária para correção das alterações faciais.
