Urea Urea A compound formed in the liver from ammonia produced by the deamination of amino acids. It is the principal end product of protein catabolism and constitutes about one half of the total urinary solids. Urea Cycle cycle disorders (UCDs) are caused by genetic defects and result in deficiencies of enzymes Enzymes Enzymes are complex protein biocatalysts that accelerate chemical reactions without being consumed by them. Due to the body's constant metabolic needs, the absence of enzymes would make life unsustainable, as reactions would occur too slowly without these molecules. Basics of Enzymes and transporters of the urea Urea A compound formed in the liver from ammonia produced by the deamination of amino acids. It is the principal end product of protein catabolism and constitutes about one half of the total urinary solids. Urea Cycle cycle. As a result of the defects, individuals are unable to rid the body of nitrogen Nitrogen An element with the atomic symbol n, atomic number 7, and atomic weight [14. 00643; 14. 00728]. Nitrogen exists as a diatomic gas and makes up about 78% of the earth's atmosphere by volume. It is a constituent of proteins and nucleic acids and found in all living cells. Urea Cycle waste. Common symptoms include vomiting Vomiting The forcible expulsion of the contents of the stomach through the mouth. Hypokalemia, lethargy Lethargy A general state of sluggishness, listless, or uninterested, with being tired, and having difficulty concentrating and doing simple tasks. It may be related to depression or drug addiction. Hyponatremia, seizures Seizures A seizure is abnormal electrical activity of the neurons in the cerebral cortex that can manifest in numerous ways depending on the region of the brain affected. Seizures consist of a sudden imbalance that occurs between the excitatory and inhibitory signals in cortical neurons, creating a net excitation. The 2 major classes of seizures are focal and generalized. Seizures, and respiratory alkalosis Alkalosis A pathological condition that removes acid or adds base to the body fluids. Respiratory Alkalosis. Most defects are autosomal recessive Autosomal recessive Autosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal recessive diseases are only expressed when 2 copies of the recessive allele are inherited. Autosomal Recessive and Autosomal Dominant Inheritance and definitive diagnosis is by molecular genetic testing Genetic Testing Detection of a mutation; genotype; karyotype; or specific alleles associated with genetic traits, heritable diseases, or predisposition to a disease, or that may lead to the disease in descendants. It includes prenatal genetic testing. Myotonic Dystrophies. Treatment aims to reduce ammonia Ammonia A colorless alkaline gas. It is formed in the body during decomposition of organic materials during a large number of metabolically important reactions. Note that the aqueous form of ammonia is referred to as ammonium hydroxide. Acid-Base Balance concentration in plasma Plasma The residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation. Transfusion Products. In less severe cases, acute episodes can be prevented through dietary restriction of protein. Untreated disease may lead to seizures Seizures A seizure is abnormal electrical activity of the neurons in the cerebral cortex that can manifest in numerous ways depending on the region of the brain affected. Seizures consist of a sudden imbalance that occurs between the excitatory and inhibitory signals in cortical neurons, creating a net excitation. The 2 major classes of seizures are focal and generalized. Seizures, coma Coma Coma is defined as a deep state of unarousable unresponsiveness, characterized by a score of 3 points on the GCS. A comatose state can be caused by a multitude of conditions, making the precise epidemiology and prognosis of coma difficult to determine. Coma, or death.
Last updated: Apr 17, 2025
Urea Urea A compound formed in the liver from ammonia produced by the deamination of amino acids. It is the principal end product of protein catabolism and constitutes about one half of the total urinary solids. Urea Cycle cycle disorders (UCDs) are a group of syndromes resulting from genetic mutations Genetic Mutations Carcinogenesis, which cause deficiencies in enzymes Enzymes Enzymes are complex protein biocatalysts that accelerate chemical reactions without being consumed by them. Due to the body’s constant metabolic needs, the absence of enzymes would make life unsustainable, as reactions would occur too slowly without these molecules. Basics of Enzymes and amino acid Amino acid Amino acids (AAs) are composed of a central carbon atom attached to a carboxyl group, an amino group, a hydrogen atom, and a side chain (R group). Basics of Amino Acids transporters of the urea Urea A compound formed in the liver from ammonia produced by the deamination of amino acids. It is the principal end product of protein catabolism and constitutes about one half of the total urinary solids. Urea Cycle cycle, resulting in the accumulation of nitrogenous waste products.
All UCDs stem from genetic abnormalities, which cause deficiencies in enzymes Enzymes Enzymes are complex protein biocatalysts that accelerate chemical reactions without being consumed by them. Due to the body’s constant metabolic needs, the absence of enzymes would make life unsustainable, as reactions would occur too slowly without these molecules. Basics of Enzymes important to the urea Urea A compound formed in the liver from ammonia produced by the deamination of amino acids. It is the principal end product of protein catabolism and constitutes about one half of the total urinary solids. Urea Cycle cycle:
All UCDs are inherited as autosomal recessive Autosomal recessive Autosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal recessive diseases are only expressed when 2 copies of the recessive allele are inherited. Autosomal Recessive and Autosomal Dominant Inheritance traits except OTC deficiency ( X-linked recessive X-Linked Recessive Duchenne Muscular Dystrophy trait).
Normal physiology of urea Urea A compound formed in the liver from ammonia produced by the deamination of amino acids. It is the principal end product of protein catabolism and constitutes about one half of the total urinary solids. Urea Cycle cycle:
Impairment of the urea Urea A compound formed in the liver from ammonia produced by the deamination of amino acids. It is the principal end product of protein catabolism and constitutes about one half of the total urinary solids. Urea Cycle cycle in the liver Liver The liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy causes hyperammonemia Hyperammonemia Elevated level of ammonia in the blood. It is a sign of defective catabolism of amino acids or ammonia to urea. Cirrhosis ( ammonia Ammonia A colorless alkaline gas. It is formed in the body during decomposition of organic materials during a large number of metabolically important reactions. Note that the aqueous form of ammonia is referred to as ammonium hydroxide. Acid-Base Balance accumulation in the blood). Depending on the severity and age at manifestation, ammonia Ammonia A colorless alkaline gas. It is formed in the body during decomposition of organic materials during a large number of metabolically important reactions. Note that the aqueous form of ammonia is referred to as ammonium hydroxide. Acid-Base Balance may cause neurotoxic effects on the brain Brain The part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem. Nervous System: Anatomy, Structure, and Classification.
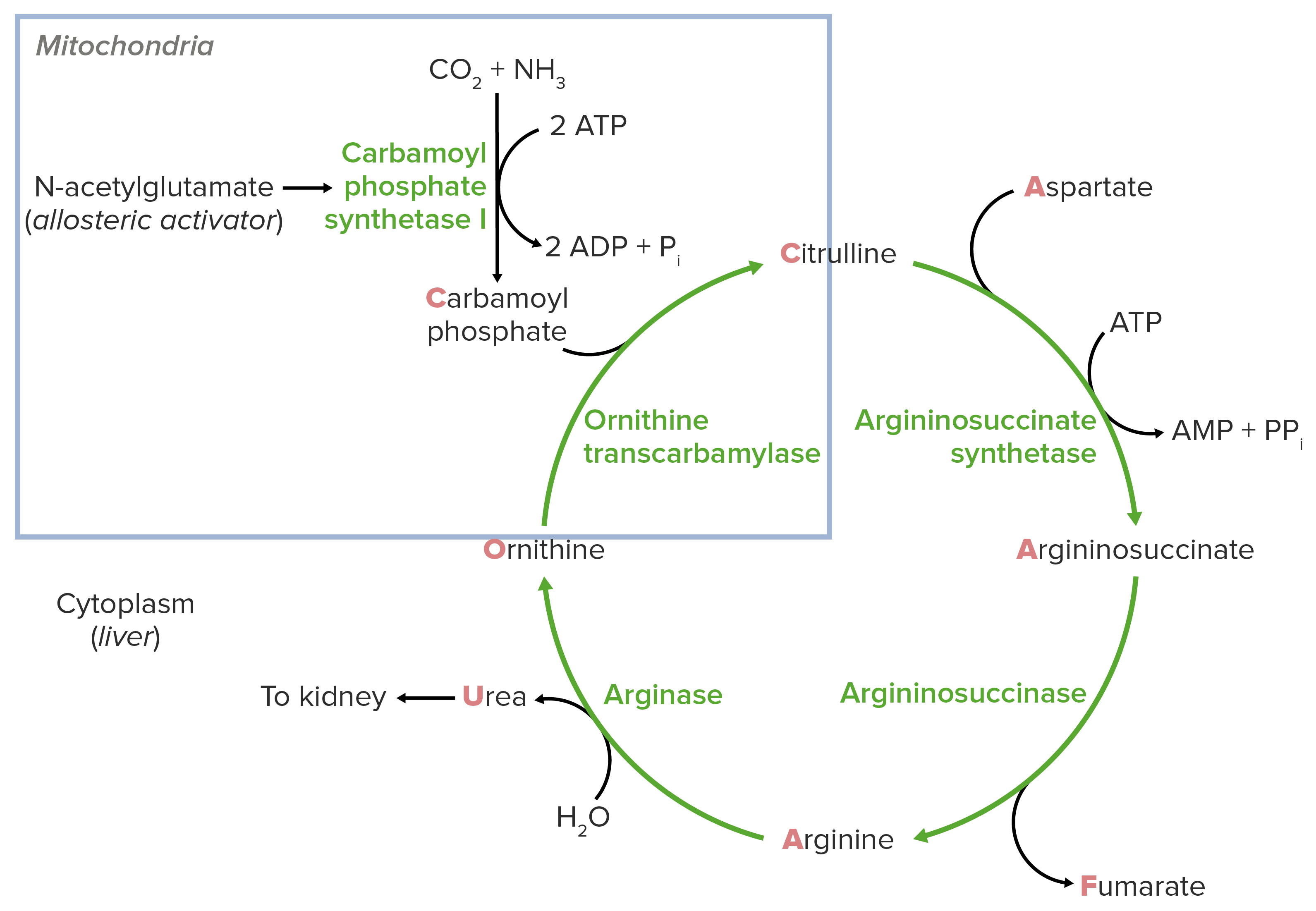
Schematic diagram of the urea cycle: The blue square indicates the feeder reaction.
Image by Lecturio.Treatment should be tailored to the specific disorder:
Untreated hyperammonemia Hyperammonemia Elevated level of ammonia in the blood. It is a sign of defective catabolism of amino acids or ammonia to urea. Cirrhosis may cause: