Los LOS Neisseria trastornos del ciclo de la urea Urea A compound formed in the liver from ammonia produced by the deamination of amino acids. It is the principal end product of protein catabolism and constitutes about one half of the total urinary solids. Urea Cycle son causados por defectos genéticos y resultan en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum deficiencias de enzimas y transportadores del ciclo de la urea Urea A compound formed in the liver from ammonia produced by the deamination of amino acids. It is the principal end product of protein catabolism and constitutes about one half of the total urinary solids. Urea Cycle. Como resultado de los LOS Neisseria defectos, los LOS Neisseria pacientes no pueden eliminar los LOS Neisseria desechos nitrogenados del cuerpo. Los LOS Neisseria síntomas comunes incluyen vómitos, letargo, convulsiones y alcalosis respiratoria. La mayoría de los LOS Neisseria defectos son autosómicos recesivos y el diagnóstico definitivo se realiza mediante pruebas genéticas moleculares. El objetivo del tratamiento es reducir la concentración de amoníaco en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el plasma Plasma The residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation. Transfusion Products. En EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum casos menos graves, los LOS Neisseria episodios agudos se pueden prevenir mediante la restricción dietética de proteínas. Si la enfermedad no es tratada puede provocar convulsiones, coma Coma Coma is defined as a deep state of unarousable unresponsiveness, characterized by a score of 3 points on the GCS. A comatose state can be caused by a multitude of conditions, making the precise epidemiology and prognosis of coma difficult to determine. Coma o la muerte.
Last updated: Apr 17, 2025
Los LOS Neisseria trastornos del ciclo de la urea Urea A compound formed in the liver from ammonia produced by the deamination of amino acids. It is the principal end product of protein catabolism and constitutes about one half of the total urinary solids. Urea Cycle son un grupo de síndromes resultantes de mutaciones genéticas que causan deficiencias en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum las enzimas y los LOS Neisseria transportadores de los LOS Neisseria aminoácidos del ciclo de la urea Urea A compound formed in the liver from ammonia produced by the deamination of amino acids. It is the principal end product of protein catabolism and constitutes about one half of the total urinary solids. Urea Cycle, lo que resulta en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la acumulación de productos de desecho nitrogenados.
Todos los LOS Neisseria trastornos del ciclo de la urea Urea A compound formed in the liver from ammonia produced by the deamination of amino acids. It is the principal end product of protein catabolism and constitutes about one half of the total urinary solids. Urea Cycle provienen de anomalías genéticas, que causan deficiencias en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum enzimas importantes para el ciclo de la urea Urea A compound formed in the liver from ammonia produced by the deamination of amino acids. It is the principal end product of protein catabolism and constitutes about one half of the total urinary solids. Urea Cycle:
Todos los LOS Neisseria trastornos del ciclo de la urea Urea A compound formed in the liver from ammonia produced by the deamination of amino acids. It is the principal end product of protein catabolism and constitutes about one half of the total urinary solids. Urea Cycle se heredan como rasgos autosómicos recesivos, excepto la deficiencia de ornitina transcarbamilasa (rasgo recesivo ligado al AL Amyloidosis cromosoma X).
Fisiología normal del ciclo de la urea Urea A compound formed in the liver from ammonia produced by the deamination of amino acids. It is the principal end product of protein catabolism and constitutes about one half of the total urinary solids. Urea Cycle:
El deterioro del ciclo de la urea Urea A compound formed in the liver from ammonia produced by the deamination of amino acids. It is the principal end product of protein catabolism and constitutes about one half of the total urinary solids. Urea Cycle en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el hígado provoca hiperamonemia (acumulación de amoníaco en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la sangre). Según la gravedad y la edad de manifestación, el amoníaco puede causar efectos neurotóxicos en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el cerebro.
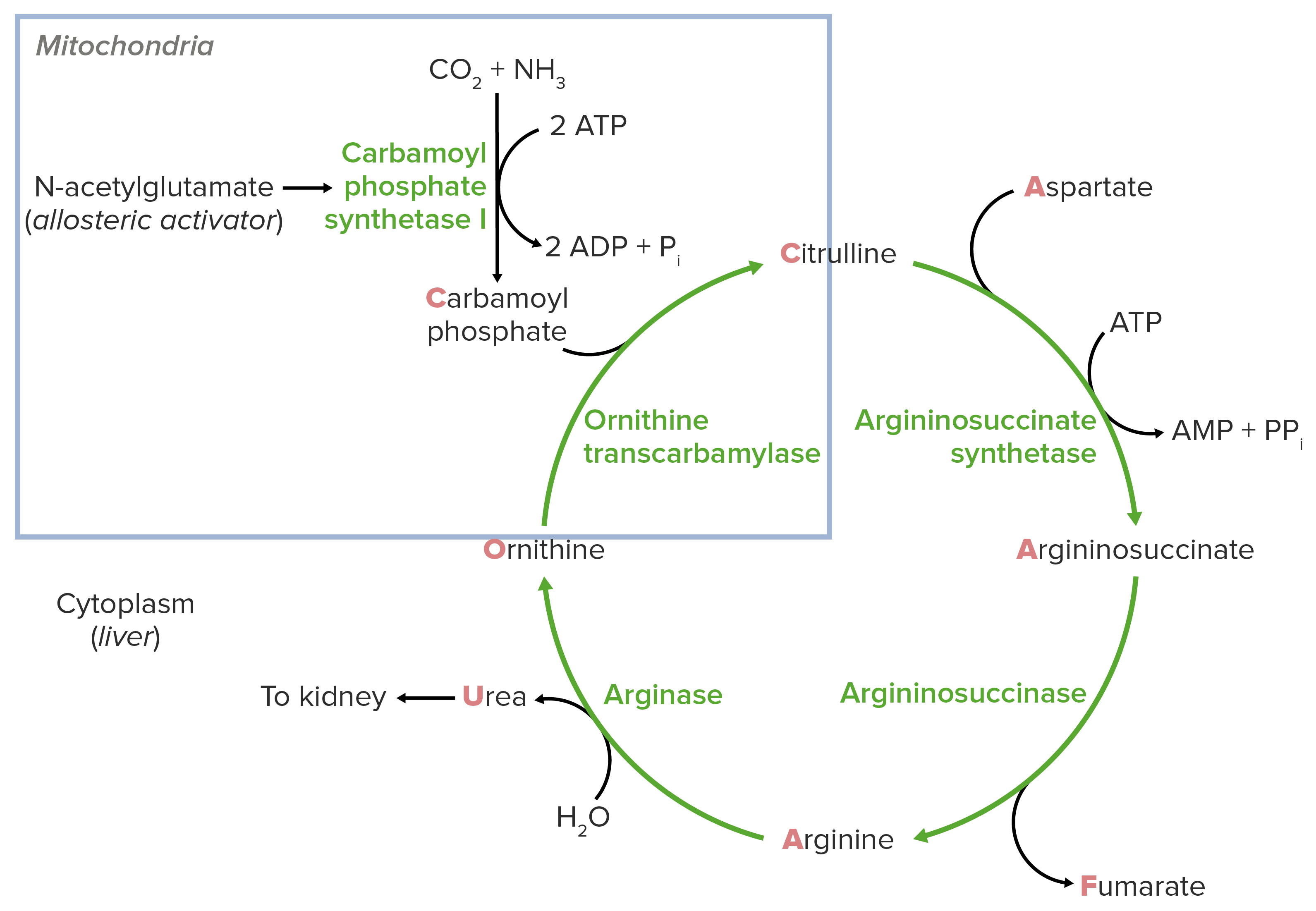
Diagrama esquemático del ciclo de la urea: el cuadrado azul indica la reacción alimentadora
Imagen por Lecturio.El tratamiento debe adaptarse al AL Amyloidosis trastorno específico:
La hiperamonemia no tratada puede causar: