


As perturbações do ciclo da ureia (UCD) são causadas por defeitos genéticos e resultam em deficiências das enzimas e dos transportadores do ciclo da ureia. Como resultado desses defeitos, os indivíduos são incapazes de livrar o corpo dos resíduos de azoto. Os sintomas comuns incluem vómitos, letargia, convulsões e alcalose respiratória. A maioria dos defeitos são autossómicos recessivos e o diagnóstico definitivo é feito através de testes Testes Gonadal Hormones genéticos moleculares. O tratamento visa reduzir a concentração de amónia no plasma Plasma The residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation. Transfusion Products. Em casos menos graves, os episódios agudos podem ser evitados através da restrição alimentar de proteínas. A doença não tratada pode levar a convulsões, coma Coma Coma is defined as a deep state of unarousable unresponsiveness, characterized by a score of 3 points on the GCS. A comatose state can be caused by a multitude of conditions, making the precise epidemiology and prognosis of coma difficult to determine. Coma ou morte.
Last updated: Apr 17, 2025
As perturbações do ciclo da ureia (UCD pela sigla em inglês) são um conjunto de síndromes resultantes de mutações genéticas, que causam deficiências nas enzimas e nos transportadores de aminoácidos do ciclo da ureia, resultando numa acumulação de produtos residuais azotados.
Todas as UCD derivam de anormalidades genéticas, que causam deficiências em enzimas importantes para o ciclo da ureia:
Todos as UCD são herdadas como características autossómicas recessivas, exceto a deficiência e OTC (característica recessiva ligada ao cromossoma X).
Fisiologia normal do ciclo da ureia:
A deficiência do ciclo da ureia no fígado causa hiperamonemia (acumulação de amónia no sangue). Dependendo da gravidade e da idade da manifestação, a amónia pode causar efeitos neurotóxicos no cérebro.
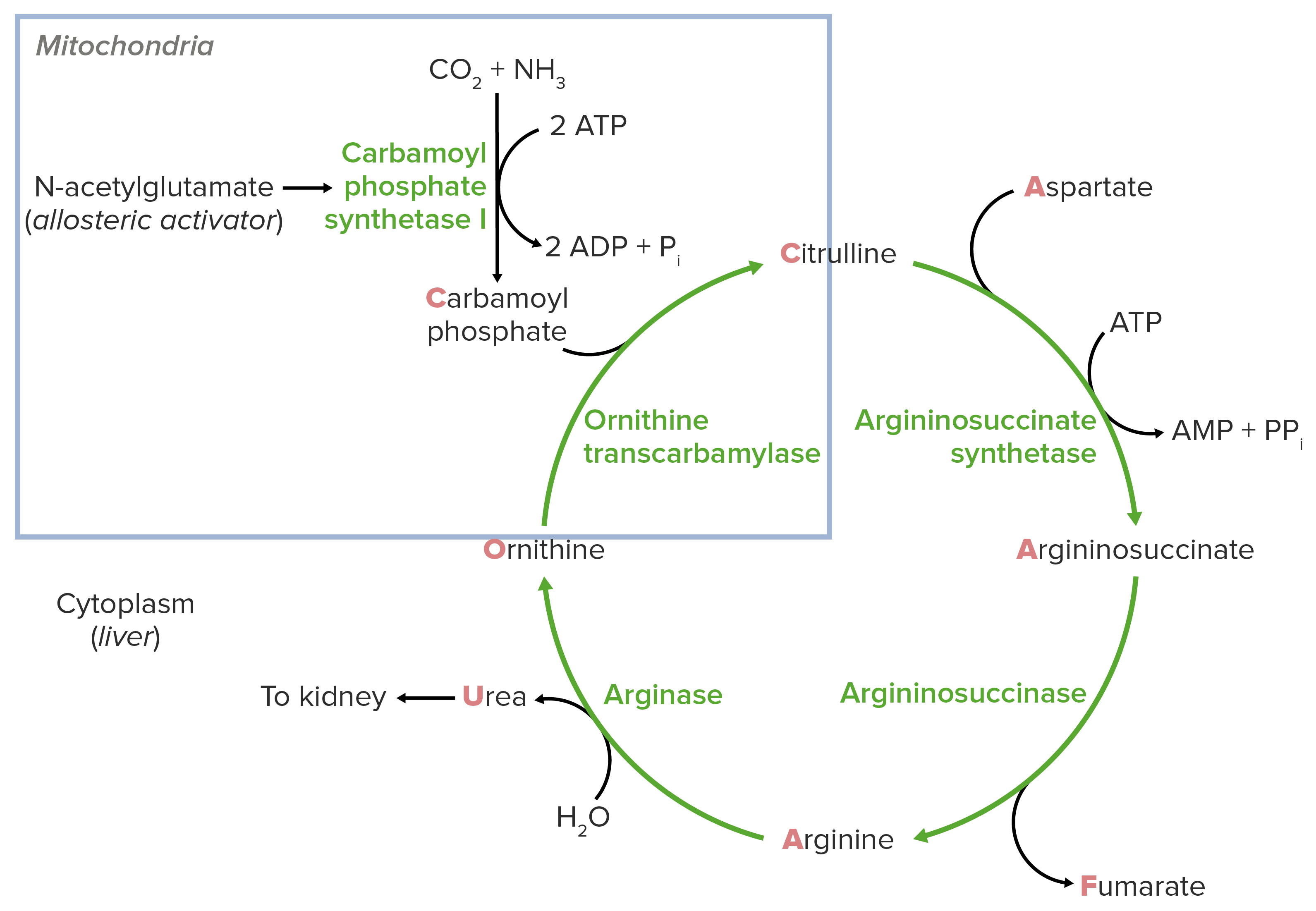
Diagrama esquemático do ciclo da ureia: O quadrado azul indica a reação “alimentadora”.
Imagem por Lecturio.O tratamento deve ser adaptado à perturbação específica:
A hiperamonemia não tratada pode causar:
