Atypical parkinsonian syndromes are a group of neurodegenerative disorders characterized by parkinsonian features although with different pathophysiology. These characteristics include progressive supranuclear palsy Palsy paralysis of an area of the body, thus incapable of voluntary movement Cranial Nerve Palsies (PSP), multiple system atrophy Atrophy Decrease in the size of a cell, tissue, organ, or multiple organs, associated with a variety of pathological conditions such as abnormal cellular changes, ischemia, malnutrition, or hormonal changes. Cellular Adaptation (MSA), and corticobasal degeneration (CBD). Although each syndrome has its own distinctive features, common factors are a rarity. There is a general propensity to affect middle-aged and elderly populations and a lack of definitive management options.
Last updated: Dec 15, 2025
Progressive supranuclear palsy Palsy paralysis of an area of the body, thus incapable of voluntary movement Cranial Nerve Palsies (PSP; also known as a Parkinson-plus disorder) is a degenerative movement disorder that affects the brain Brain The part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem. Nervous System: Anatomy, Structure, and Classification stem, basal ganglia Basal Ganglia Basal ganglia are a group of subcortical nuclear agglomerations involved in movement, and are located deep to the cerebral hemispheres. Basal ganglia include the striatum (caudate nucleus and putamen), globus pallidus, substantia nigra, and subthalamic nucleus. Basal Ganglia: Anatomy, diencephalon Diencephalon The paired caudal parts of the prosencephalon from which the thalamus; hypothalamus; epithalamus; and subthalamus are derived. Development of the Nervous System and Face, and cortex, causing gaze dysfunction, extrapyramidal symptoms Extrapyramidal Symptoms Ataxia-telangiectasia, and cognitive dysfunction.
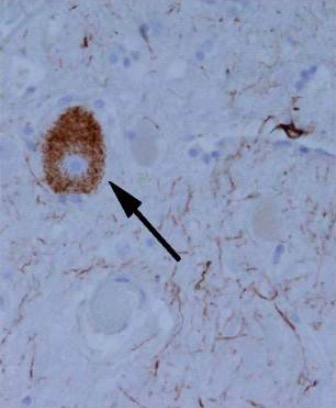
Tangle-like tau protein aggregate in an individual with progressive supranuclear palsy
Image: “Tangle-like tau protein aggregate in a patient with progressive supranuclear palsy” by Ling et al. License: CC BY 2.0, cropped by Lecturio.History:
Physical examination:
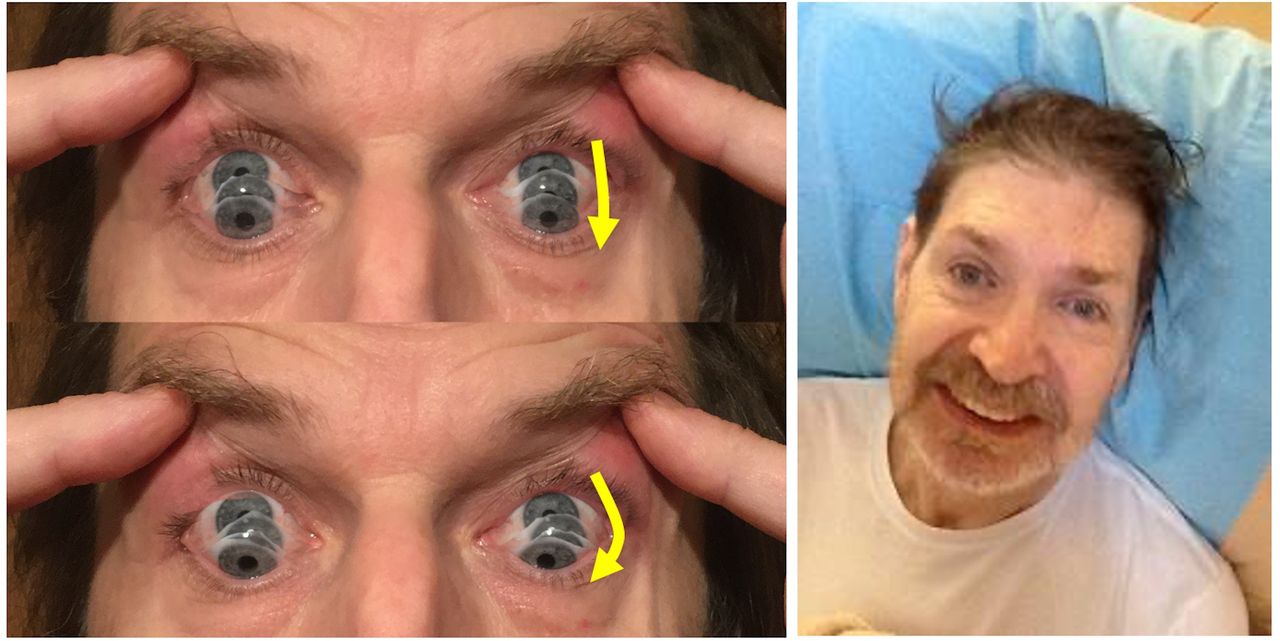
Left: The photographs show the “round-the-houses” sign, illustrated for a downward saccade. Note the lateral curvature of downward path of eye movement (yellow arrows). The velocity may also be slow.
Right: This face gives many clues to the diagnosis. As shown by this man, there may be retrocollis, raised eyebrows, and frontalis overactivity. People with progressive supranuclear palsy may have a rather fixed smile, with lips drawn back rather than up.
Initial diagnosis requires fulfillment of diagnostic criteria for PSP. Definitive diagnosis can only be made via pathologic examination.
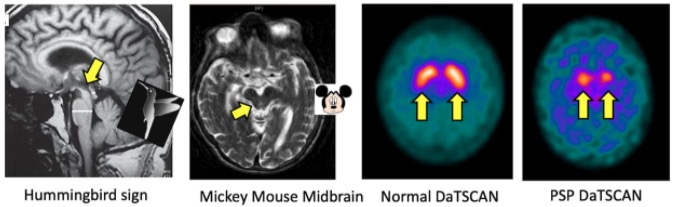
MRI and DaTscan (nuclear imaging test that allows doctors to view the brain’s dopamine levels) features in progressive supranuclear palsy (PSP)
Image: “MRI and DaTscan features in progressive supranuclear palsy” by James B. Rowe, University of Cambridge Department of Clinical Neurosciences, Cambridge, Cambridgeshire, UK. License: CC BY 4.0Multiple system atrophy Atrophy Decrease in the size of a cell, tissue, organ, or multiple organs, associated with a variety of pathological conditions such as abnormal cellular changes, ischemia, malnutrition, or hormonal changes. Cellular Adaptation (MSA) is an adult-onset, sporadic Sporadic Selective IgA Deficiency, rapidly progressive, multisystem, fatal neurodegenerative disease characterized by parkinsonian features, cerebellar, autonomic, and urogenital dysfunction; and corticospinal disorders.
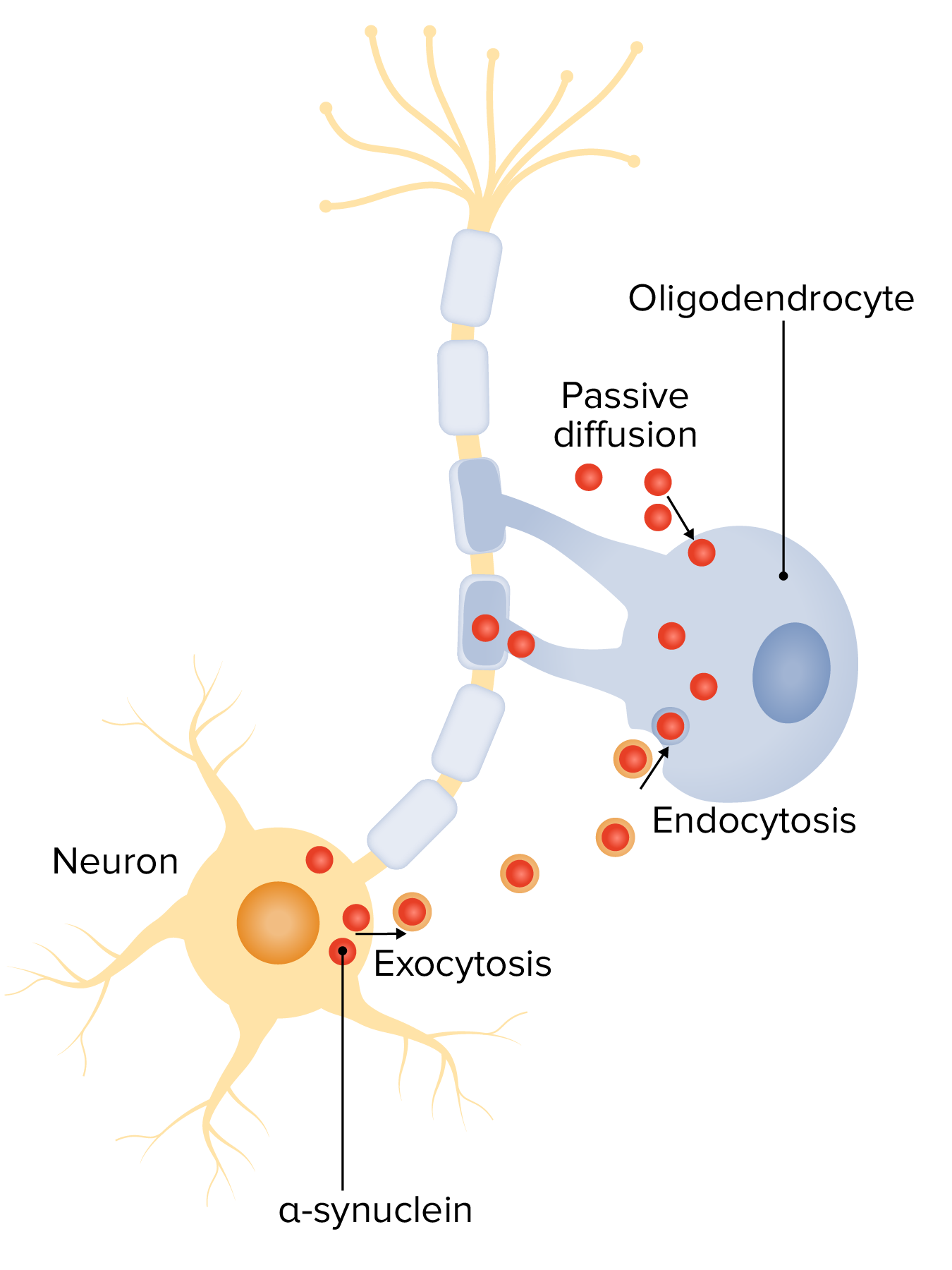
Alpha-synuclein in the pathogenesis of multiple system atrophy (MSA):
Neuron-oligodendrocyte interaction mechanisms potentially involved in α-syn accumulation: oligodendroglial α-syn uptake from surrounding neurons and extracellular environment through endocytosis and passive transmembrane diffusion
History:
Physical examination:
Diagnosis is made clinically, and definitive diagnosis is done on postmortem pathologic examination.
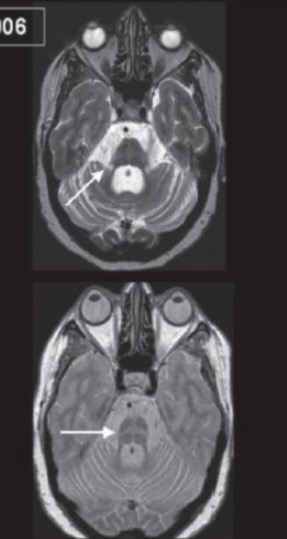
MRI of a brain showing multiple system atrophy (MSA):
Note the brain stem and cerebellar atrophy (top, arrow), and the “hot cross bun” sign (bottom, arrow).
Corticobasal degeneration (CBD) is a progressive, asymmetric dementia Dementia Major neurocognitive disorders (NCD), also known as dementia, are a group of diseases characterized by decline in a person’s memory and executive function. These disorders are progressive and persistent diseases that are the leading cause of disability among elderly people worldwide. Major Neurocognitive Disorders–movement disorder with sporadic Sporadic Selective IgA Deficiency neurodegenerative 4-repeat tauopathy.
History:
Physical examination:
Diagnosis is made clinically, and definitive diagnosis is by postmortem pathology examination.
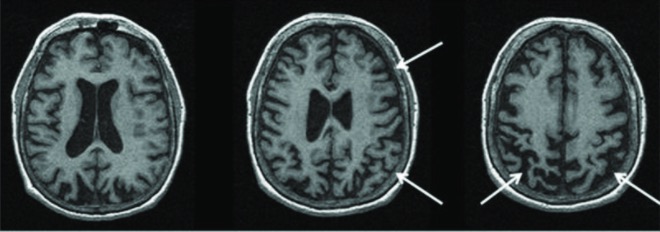
MRI of a brain showing a pathologic diagnosis of corticobasal degeneration:
MRI shows the right greater than left parietofrontal atrophy typical of corticobasal syndrome.