Los LOS Neisseria síndromes parkinsonianos atípicos son un grupo de trastornos neurodegenerativos caracterizados por rasgos parkinsonianos aunque con diferente fisiopatología. Estos trastornos incluyen parálisis supranuclear progresiva, atrofia multisistémica y degeneración corticobasal. Aunque cada síndrome tiene sus propias características distintivas, los LOS Neisseria factores comunes son una rareza. Existe una propensión general a afectar a poblaciones de mediana edad y ancianas y una falta de opciones de tratamiento definitivas.
Last updated: Dec 15, 2025
La parálisis supranuclear progresiva (también conocida como trastorno de Parkinson plus) es un trastorno degenerativo del movimiento que afecta el tronco encefálico, los LOS Neisseria ganglios basales, el diencéfalo y la corteza, causando disfunción de la mirada, síntomas extrapiramidales y disfunción cognitiva.
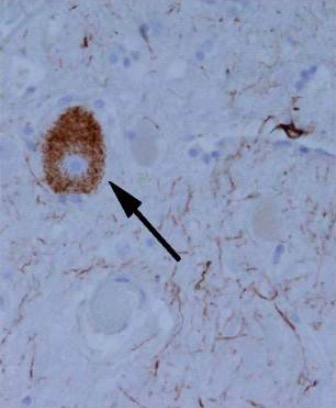
Maraña de proteína tau en forma de agregado en un individuo con parálisis supranuclear progresiva
Imagen: “Tangle-like tau protein aggregate in a patient with progressive supranuclear palsy” por Ling et al. Licencia: CC BY 2.0 , recortada por Lecturio.Antecedentes:
Examen físico:
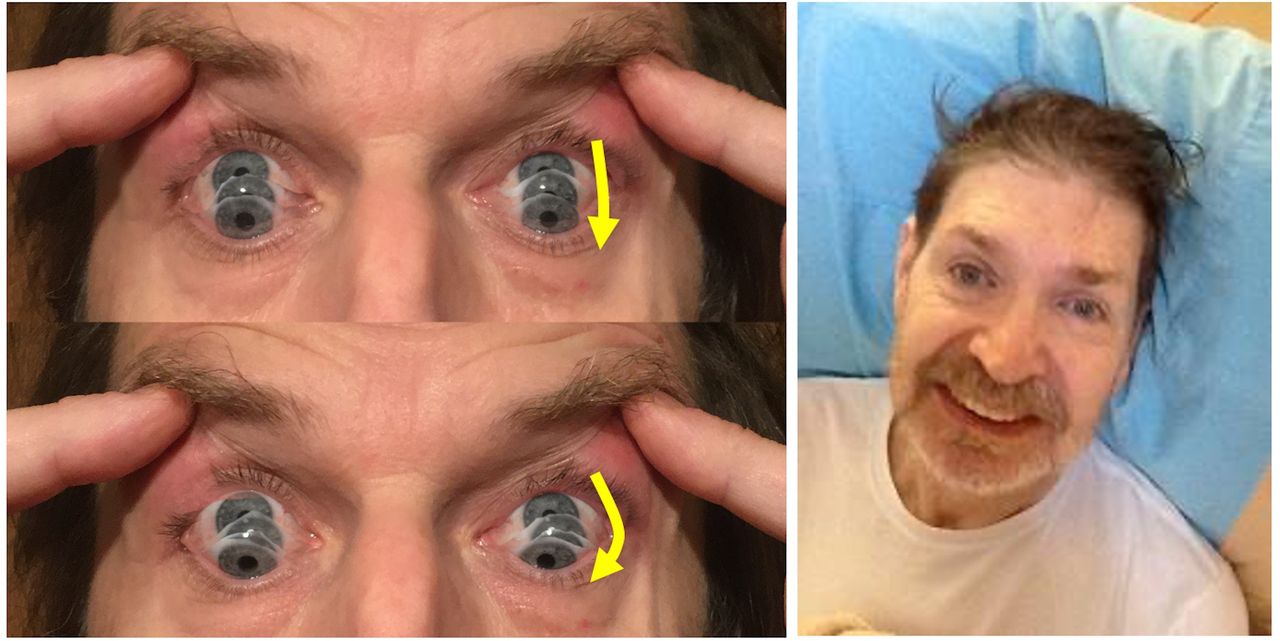
Izquierda: Las fotografías muestran el signo de “roda casas”, ilustrado para un movimiento sacádico hacia abajo. Se debe considerar la curvatura lateral de la trayectoria descendente del movimiento ocular (flechas amarillas). La velocidad también puede ser lenta.
Derecha: esta fascie da muchas pistas para el diagnóstico. Como lo muestra este hombre, puede haber retrocolis, elevación de las cejas e hiperactividad frontal. Las personas con parálisis supranuclear progresiva pueden tener una sonrisa bastante fija, con los labios hacia atrás en lugar de hacia arriba.
El diagnóstico inicial requiere el cumplimiento de los LOS Neisseria criterios diagnósticos de PSP PSP A degenerative disease of the central nervous system characterized by balance difficulties; ocular motility disorders (supranuclear ophthalmoplegia); dysarthria; swallowing difficulties; and axial dystonia. Onset is usually in the fifth decade and disease progression occurs over several years. Pathologic findings include neurofibrillary degeneration and neuronal loss in the dorsal mesencephalon; subthalamic nucleus; red nucleus; pallidum; dentate nucleus; and vestibular nuclei. Atypical Parkinsonian Syndromes. El diagnóstico definitivo sólo se puede hacer a través de un examen patológico.
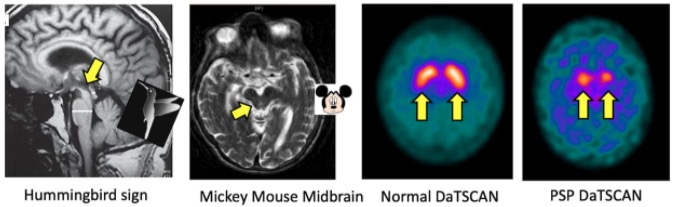
RM y DaTscan (prueba de imagen nuclear que permite a los médicos observar los niveles de dopamina del cerebro) características en la parálisis supranuclear progresiva (PSP)
Imagen: “‘MRI and DaTscan features in progressive supranuclear palsy” por James B. Rowe, University of Cambridge Department of Clinical Neurosciences, Cambridge, Cambridgeshire, UK. Licencia: CC BY 4.0La atrofia multisistémica es una enfermedad neurodegenerativa fatal, multisistémica, esporádica, rápidamente progresiva y de aparición en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum adultos caracterizada por características parkinsonianas, disfunción cerebelosa, autonómica y urogenital; y trastornos corticoespinales.
Alfa-sinucleína en la patogénesis de la atrofia multisistémica (AMS):
Mecanismos de interacción neurona-oligodendrocitos potencialmente involucrados en la acumulación de la α-syn: captación de la α-syn oligodendroglial de las neuronas circundantes y el entorno extracelular a través de endocitosis y difusión transmembranal pasiva
Antecedentes:
Examen físico:
El diagnóstico se realiza clínicamente y el diagnóstico definitivo se realiza en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el examen patológico post mortem.
RM de un cerebro que muestra atrofia multisistémica:
Obsérvese la atrofia del tronco encefálico y del cerebelo (flecha superior) y el signo del “bollo cruzado caliente” (flecha inferior).
La degeneración corticobasal es un trastorno progresivo de demencia–trastornos del movimiento asimétrico con tauopatía neurodegenerativa esporádica de 4 repeticiones.
Antecedentes:
Examen físico:
El diagnóstico se realiza clínicamente y el diagnóstico definitivo se realiza mediante un examen patológico post mortem.
RM de un cerebro que muestra un diagnóstico patológico de degeneración corticobasal:
La RM muestra la atrofia parietofrontal derecha mayor que la izquierda típica del síndrome corticobasal.