


As síndromes parkinsónicas atípicas são um grupo de doenças neurodegenerativas caracterizadas por achados parkinsónicos, embora com uma fisiopatologia diferente. Estes achados incluem paralisia supranuclear progressiva ( PSP PSP A degenerative disease of the central nervous system characterized by balance difficulties; ocular motility disorders (supranuclear ophthalmoplegia); dysarthria; swallowing difficulties; and axial dystonia. Onset is usually in the fifth decade and disease progression occurs over several years. Pathologic findings include neurofibrillary degeneration and neuronal loss in the dorsal mesencephalon; subthalamic nucleus; red nucleus; pallidum; dentate nucleus; and vestibular nuclei. Atypical Parkinsonian Syndromes), atrofia de múltiplos sistemas ( AMS AMS Neurologic syndrome with no physical findings occurring > 6 hours after ascent to altitudes > 2,500 m (rarely, 1,500 m) Altitude Sickness) e degeneração corticobasal (DCB). Embora cada síndrome tenha as suas próprias características, os fatores comuns são uma raridade. Existe uma propensão geral para afetar as populações de meia-idade e idosas e uma escassa lista de opções de tratamentos definitivos.
Last updated: Dec 15, 2025
A paralisia supranuclear progressiva ( PSP PSP A degenerative disease of the central nervous system characterized by balance difficulties; ocular motility disorders (supranuclear ophthalmoplegia); dysarthria; swallowing difficulties; and axial dystonia. Onset is usually in the fifth decade and disease progression occurs over several years. Pathologic findings include neurofibrillary degeneration and neuronal loss in the dorsal mesencephalon; subthalamic nucleus; red nucleus; pallidum; dentate nucleus; and vestibular nuclei. Atypical Parkinsonian Syndromes; também conhecida como Parkinson-plus) é um distúrbio degenerativo do movimento que afeta o tronco cerebral, os gânglios da base, o diencéfalo e o córtex, causando disfunção do olhar, sintomas extrapiramidais e disfunção cognitiva.
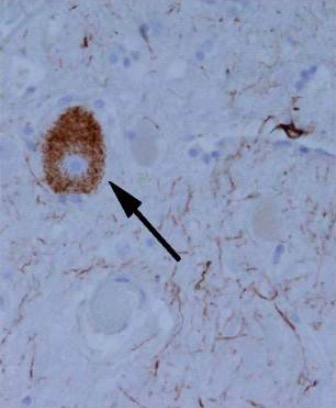
Agregados de proteína tau semelhantes a um emaranhado, num indivíduo com paralisia supranuclear progressiva
Imagem: “Tangle-like tau protein aggregate in a patient with progressive supranuclear palsy” por Ling et al. Licença: CC BY 2.0 , recortado por Lecturio.História clínica:
Exame objetivo:
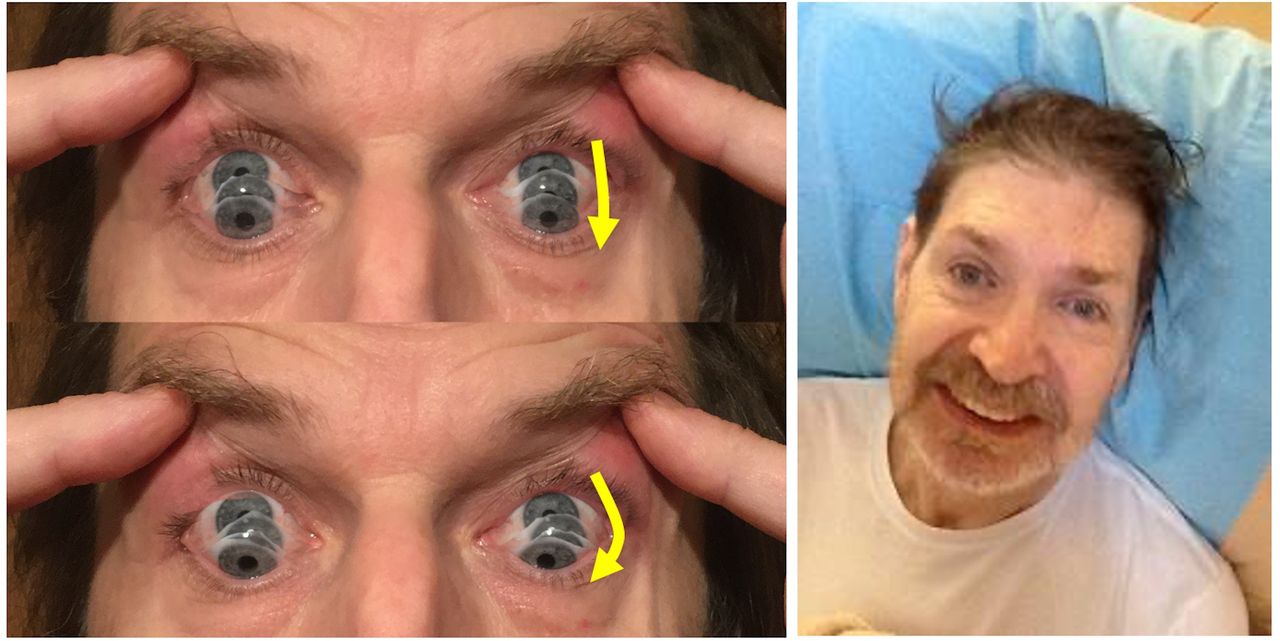
Esquerda: As fotografias mostram o sinal da sacada semilunar (round-the-houses), ilustrada por uma sacada descendente. Observar a curvatura lateral do caminho descendente do movimento dos olhos (setas amarelas). A velocidade também pode ser lenta.
À direita: este rosto fornece muitas pistas para o diagnóstico. Conforme mostrado neste homem, pode haver retrocolis, elevação das sobrancelhas e hiperatividade do frontal. Indivíduos com paralisia supranuclear progressiva podem ter um sorriso bastante fixo, com os lábios puxados para trás em vez de para cima.
O diagnóstico inicial requer o cumprimento dos critérios de diagnóstico para a PSP PSP A degenerative disease of the central nervous system characterized by balance difficulties; ocular motility disorders (supranuclear ophthalmoplegia); dysarthria; swallowing difficulties; and axial dystonia. Onset is usually in the fifth decade and disease progression occurs over several years. Pathologic findings include neurofibrillary degeneration and neuronal loss in the dorsal mesencephalon; subthalamic nucleus; red nucleus; pallidum; dentate nucleus; and vestibular nuclei. Atypical Parkinsonian Syndromes. O diagnóstico definitivo só pode ser feito por meio de exame patológico.
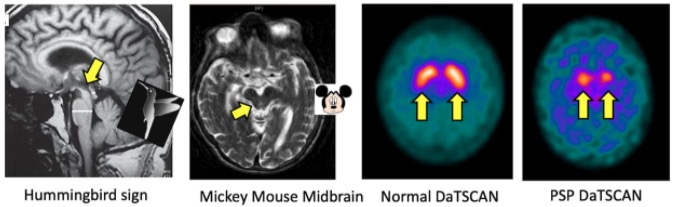
RMN e DaTscan (teste de imagem nuclear que permite aos médicos ver os níveis de dopamina do cérebro) são característicos na paralisia supranuclear progressiva (PSP)
Imagem: “‘Round-the-houses’ sign” por James B. Rowe, University of Cambridge Department of Clinical Neurosciences, Cambridge, Cambridgeshire, UK. Licença: CC BY 4.0A atrofia de múltiplos sistemas ( AMS AMS Neurologic syndrome with no physical findings occurring > 6 hours after ascent to altitudes > 2,500 m (rarely, 1,500 m) Altitude Sickness) é uma doença neurodegenerativa fatal, multissistémica, de início no adulto, esporádica, rapidamente progressiva, caracterizada por sintomas parkinsónicos, disfunção cerebelar, autonómica e urogenital; e distúrbios corticospinhais.
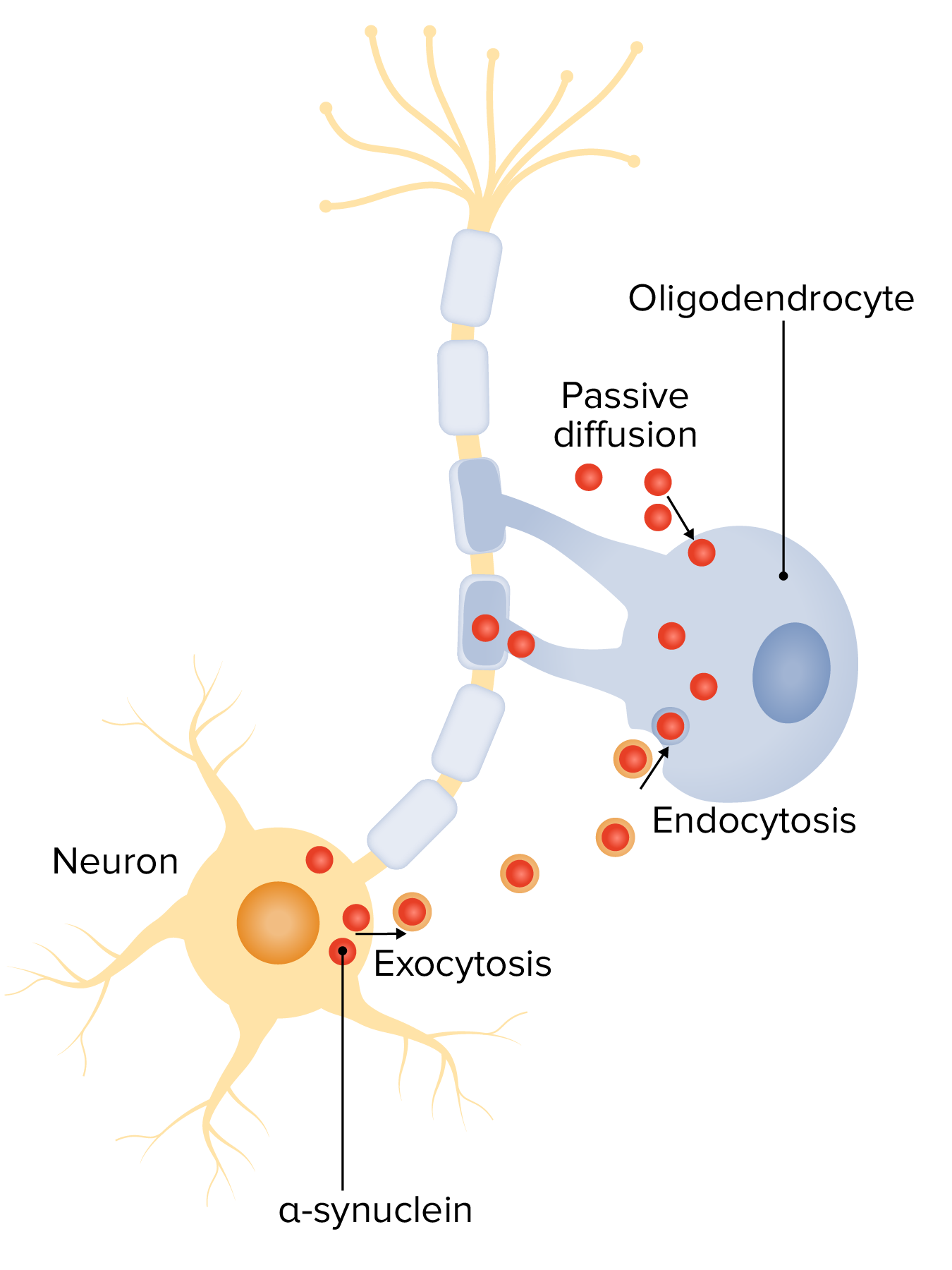
Alfa-sinucleína na patogénese da atrofia de múltiplos sistemas (AMS):
Mecanismos de interação neurónio-oligodendrócito potencialmente envolvidos na acumulação de α-syn: captação oligodendroglial de α-syn dos neurónios circundantes e ambiente extracelular, por meio de endocitose e difusão transmembranar passiva
História clínica:
Exame objetivo:
O diagnóstico clínico e o diagnóstico definitivo é baseado no exame patológico post mortem.
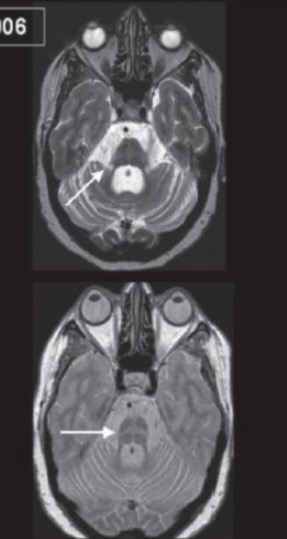
RMN de um cérebro demonstrando atrofia de múltiplos sistemas (AMS):
Observar a atrofia do tronco cerebral e do cerebelo (em cima, seta) e o sinal da cruz (embaixo, seta).
A degeneração corticobasal (DCB) é um distúrbio demencial e do movimento, progressivo e assimétrico, com tauopatia neurodegenerativa esporádica de 4 repetições.
História clínica:
Exame objetivo:
O diagnóstico é clínico e o diagnóstico definitivo é baseado no exame anatomopatológico post mortem.
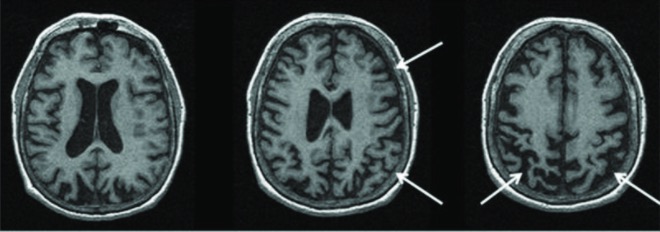
RNM de um cérebro demonstrando um diagnóstico patológico de degeneração corticobasal:
A RMN mostra atrofia parietofrontal direita maior do que a esquerda, típica da síndrome corticobasal.
