Anorectal atresia Atresia Hypoplastic Left Heart Syndrome (HLHS) refers to a spectrum of congenital anorectal malformations with an unclear etiology. These anomalies range from a simple imperforate anus in an otherwise normal anorectal region to complex anomalies involving the urogenital system. Anorectal atresia Atresia Hypoplastic Left Heart Syndrome (HLHS) is sometimes seen as an isolated finding, but it may also occur as part of a multi-organ syndrome. Diagnosis is often made during initial newborn Newborn An infant during the first 28 days after birth. Physical Examination of the Newborn examination or after a delay in passage of meconium Meconium The thick green-to-black mucilaginous material found in the intestines of a full-term fetus. It consists of secretions of the intestinal glands; bile pigments; fatty acids; amniotic fluid; and intrauterine debris. It constitutes the first stools passed by a newborn. Prenatal and Postnatal Physiology of the Neonate for over 24 hours after birth. Prognosis Prognosis A prediction of the probable outcome of a disease based on a individual's condition and the usual course of the disease as seen in similar situations. Non-Hodgkin Lymphomas varies based on complexity. Treatment is primarily surgical.
Last updated: Dec 15, 2025
Anorectal atresia Atresia Hypoplastic Left Heart Syndrome (HLHS) arises out of deviations from the normal embryological development of abdominal organs to create the adult rectum Rectum The rectum and anal canal are the most terminal parts of the lower GI tract/large intestine that form a functional unit and control defecation. Fecal continence is maintained by several important anatomic structures including rectal folds, anal valves, the sling-like puborectalis muscle, and internal and external anal sphincters. Rectum and Anal Canal: Anatomy and anal canal.
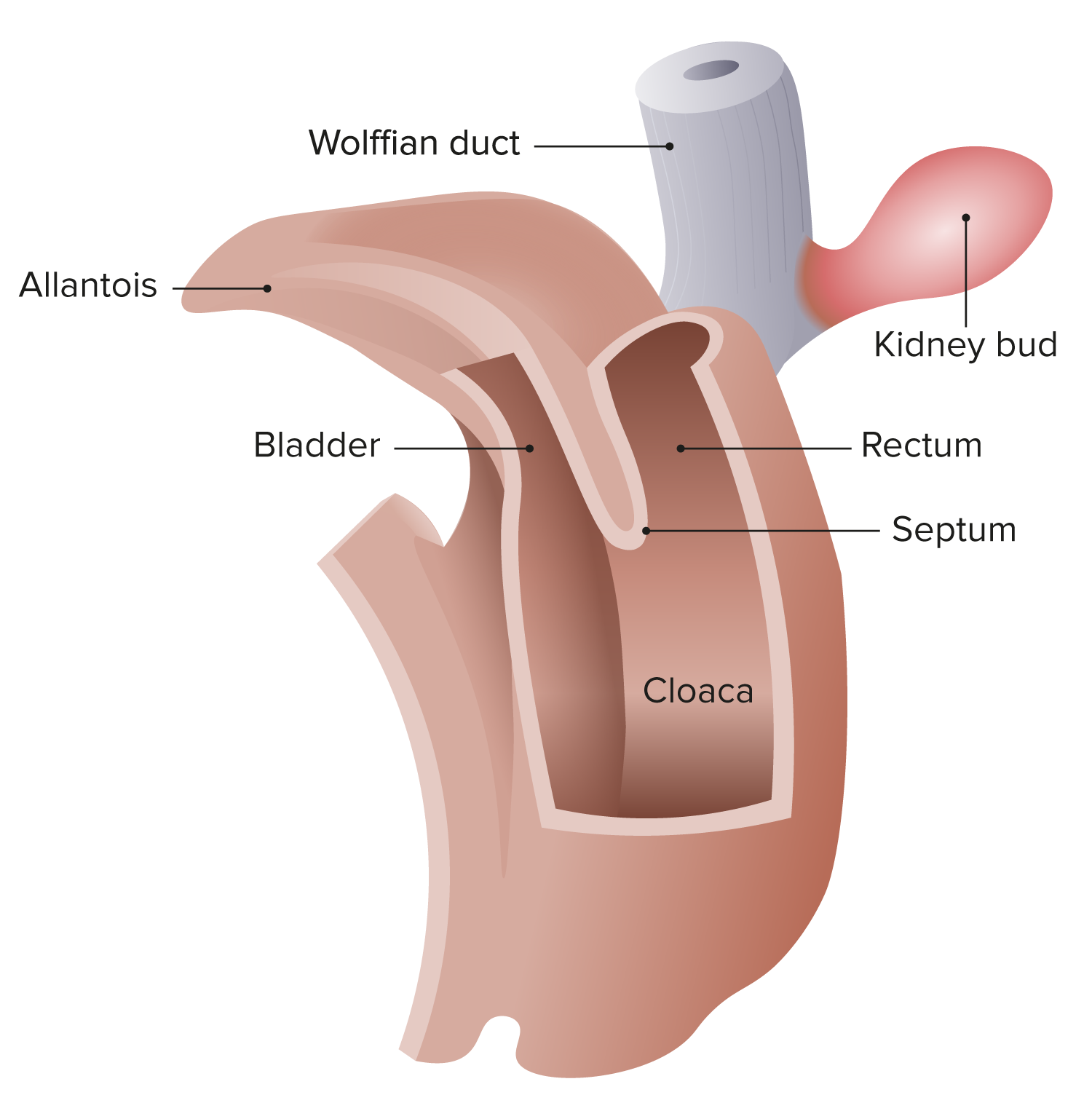
Cloaca of the human embryo at 25–27 days
Image by Lecturio.Anorectal malformations may be divided into high and low lesions based on the level of the pelvic floor Pelvic floor Soft tissue formed mainly by the pelvic diaphragm, which is composed of the two levator ani and two coccygeus muscles. The pelvic diaphragm lies just below the pelvic aperture (outlet) and separates the pelvic cavity from the perineum. It extends between the pubic bone anteriorly and the coccyx posteriorly. Vagina, Vulva, and Pelvic Floor: Anatomy (levator ani muscle):
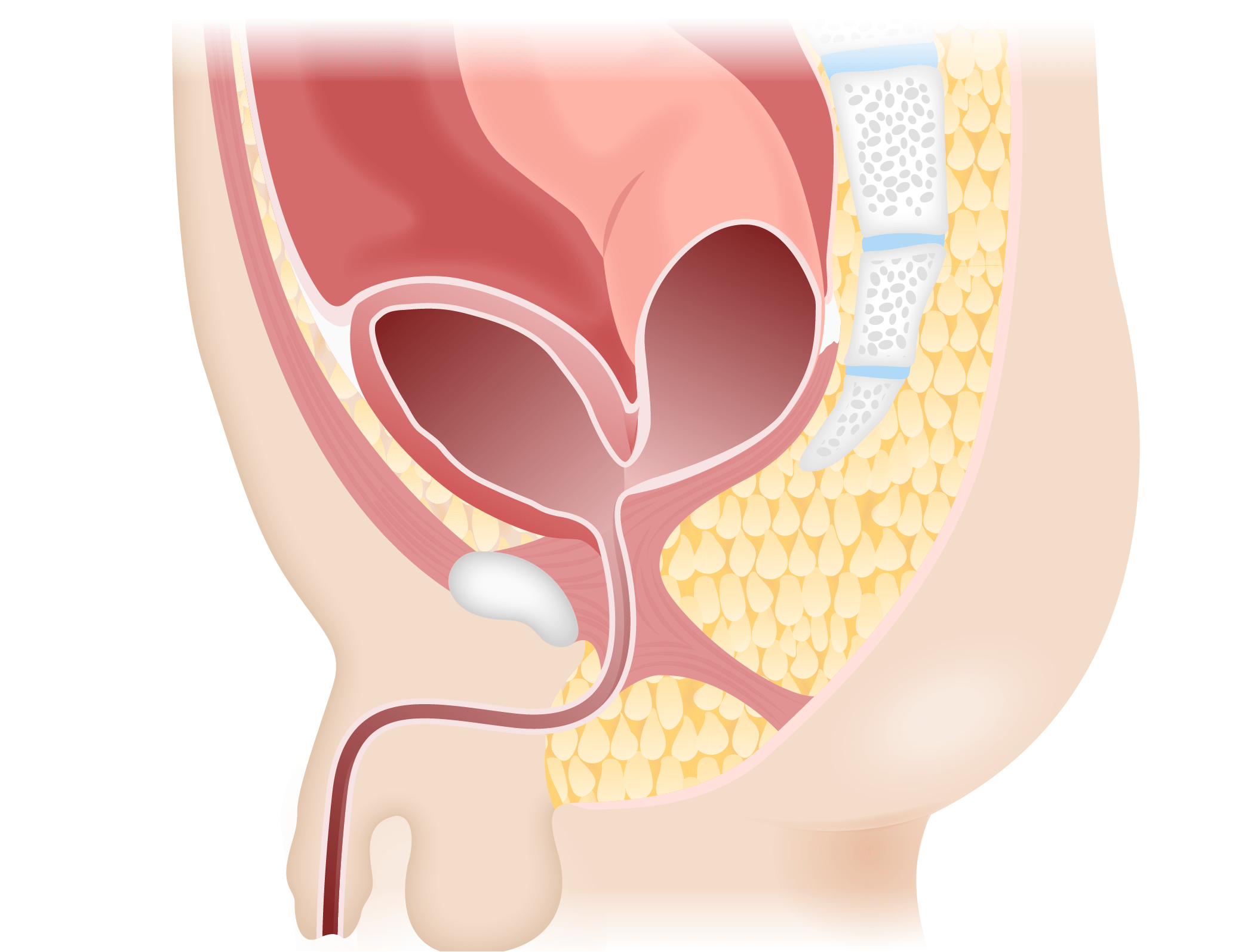
Recto-bladder neck fistula
Image by Lecturio.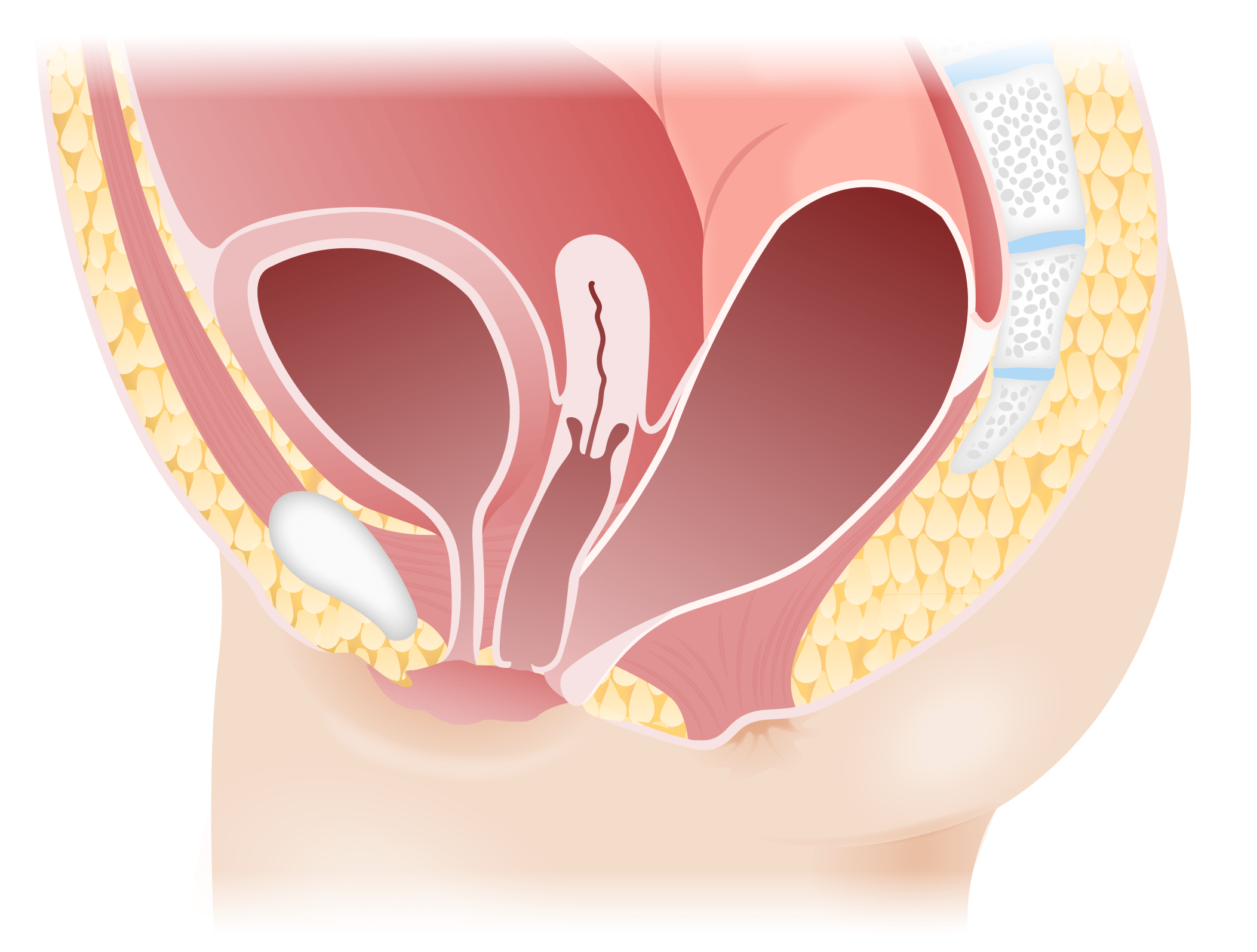
Rectovestibular fistula in females
Image by Lecturio.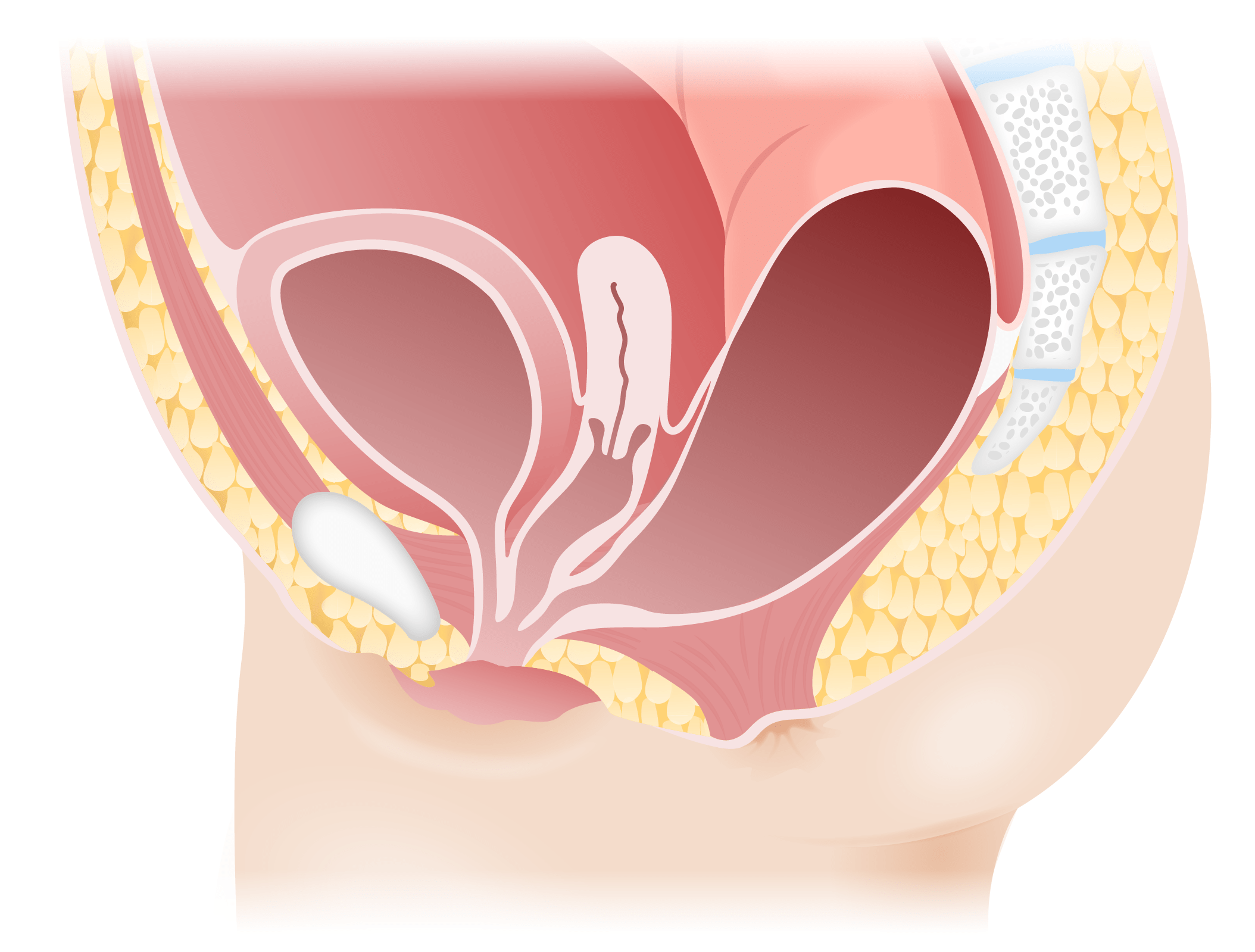
Persistent cloaca
Image by Lecturio.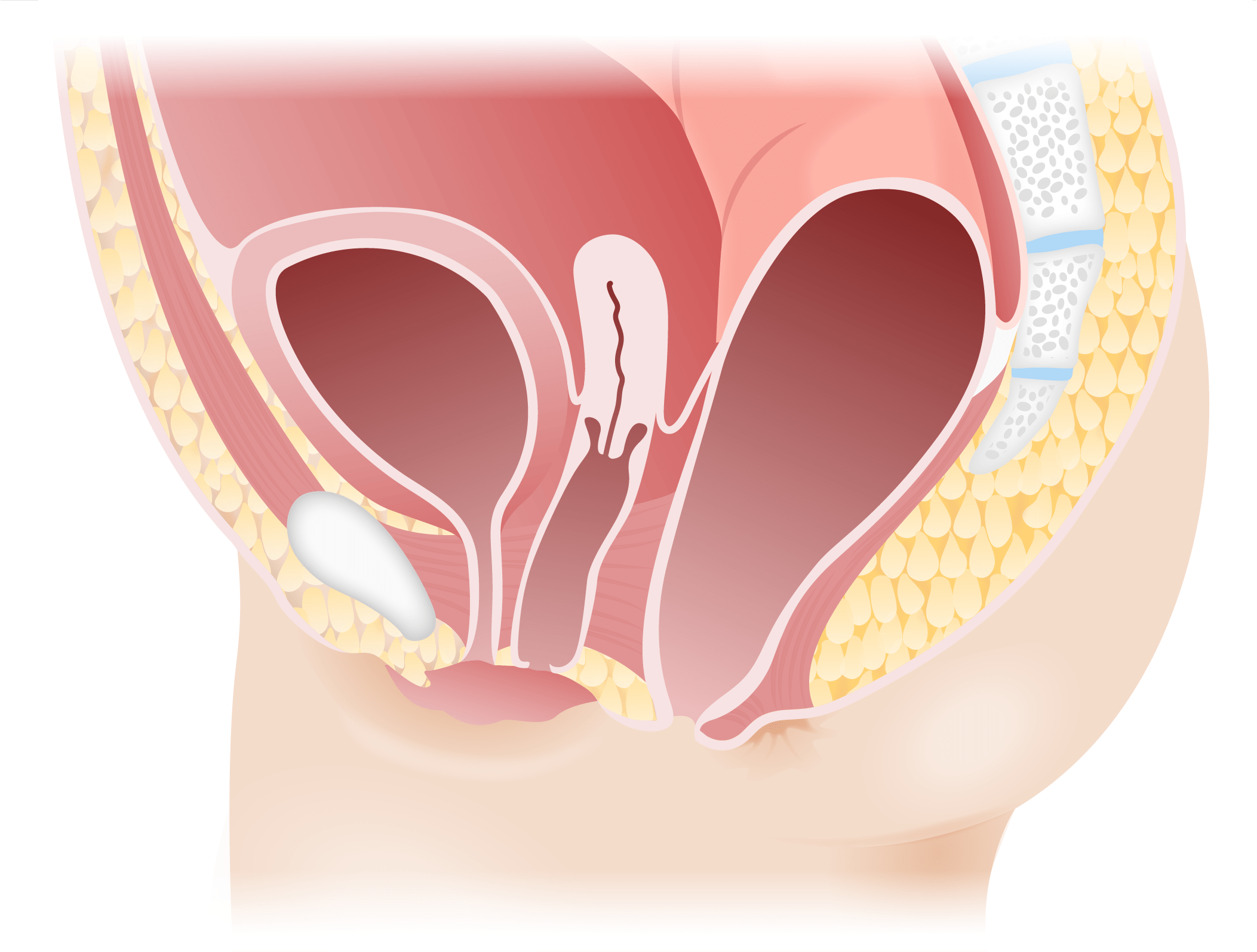
Perineal fistula
Image by Lecturio.Defects are often identified during the newborn Newborn An infant during the first 28 days after birth. Physical Examination of the Newborn physical examination. Missed cases are usually identified within 24 hours following failure to pass meconium Meconium The thick green-to-black mucilaginous material found in the intestines of a full-term fetus. It consists of secretions of the intestinal glands; bile pigments; fatty acids; amniotic fluid; and intrauterine debris. It constitutes the first stools passed by a newborn. Prenatal and Postnatal Physiology of the Neonate and abdominal distension.

Imperforate anus
Image: “Persistent cloaca perineum” by Department of Pediatric Surgery, Cincinnati Children’s Hospital, University of Cincinnati, Cincinnati, Ohio 45229, USA. License: CC BY 2.0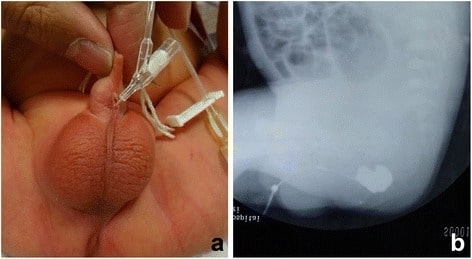
(a) A catheter was inserted into the orifice of the fistula.
(b) Fistulography showed the fistula and the end of the rectum.
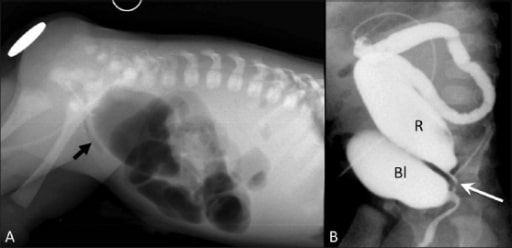
Anorectal malformation: high imperforate anus with rectouretheral fistula
Cross-table lateral radiograph (A) shows absence of air in the anal area (metallic marker at the anal verge). A lucent curvilinear air shadow is seen anteriorly (arrow), representing air in the urinary bladder as a result of the fistula.
Loopogram (B) of the same patient confirms the rectourethral fistula and outlines the rectum and the bladder.
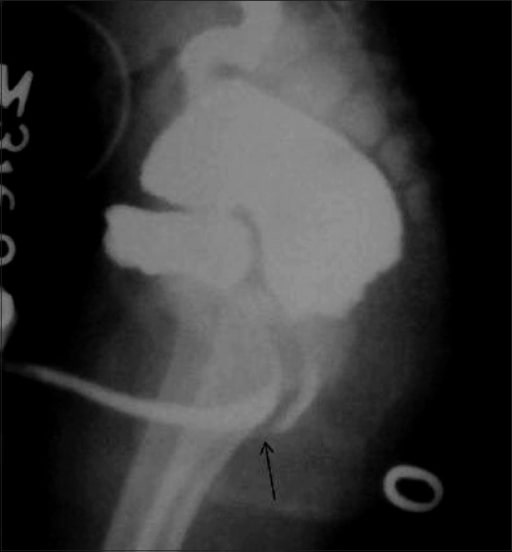
Rectobulbar fistula with anorectal atresia in an infant who presented in the early neonatal period with passage of meconium through the urethral route.
A distal cologram (performed through a transverse colostomy) reveals the absence of the distal rectum and anal canal, with communication between the rectum and the bulbar urethra through a fistula (arrow).
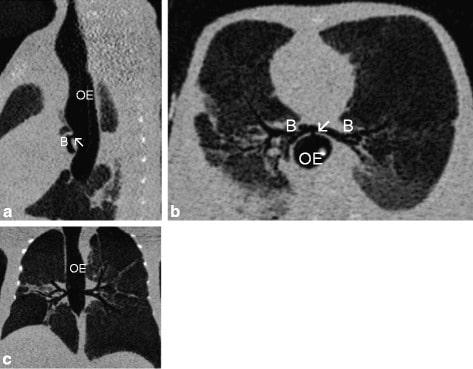
Chest computed tomography (CT) scans of newborn delivered by cesarean section to a 24-year-old mother who had polyhydramnios prenatally. Physical examination was remarkable for anal atresia, single transverse palmar crease on the right hand, and bilateral clinodactyly.
Sagittal (a), horizontal (b), and axial (c) show the absence of the trachea and bronchi (B) arising from the oesophagus (OE) via a fistula (arrow ↖).
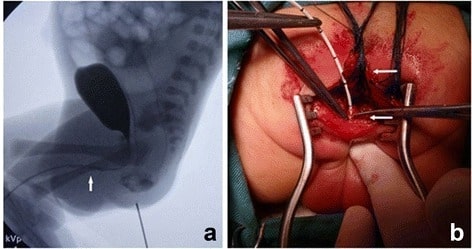
A urethrography (a) showing the urethra and bladder, with a small amount of contrast appearing in the rectum. The location of the fistula entering the urethra is displayed (arrow). Limited posterior sagittal anorectoplasty (b) with division of the rectum (upper arrow) and a catheter in the fistula (lower arrow).
Image: “Urethrography showed the urethra and the bladder” by Department of Pediatric Surgery, West China Hospital, Sichuan University, Chengdu, 610041, People’s Republic of China. License: CC BY 4.0