Long QT syndrome (LQTS) is a disorder of ventricular myocardial repolarization that produces QT prolongation on electrocardiogram (ECG). Long QT syndrome is associated with an increased risk of developing life-threatening cardiac arrhythmias, specifically torsades de pointes. The condition may be congenital or acquired. Congenital LQTS is attributed to genetic mutations affecting cardiac ion channels. Acquired LQTS usually results from drug therapy or electrolyte abnormalities. Patients can be asymptomatic or present with palpitations, syncope, seizures, and even sudden cardiac death. Diagnosis is established with ECG along with medical and family history, laboratory workup, and other cardiac tests. Treatment is determined by etiology. Acquired LQTS requires removal of the offending drug or drug combinations and correction of electrolyte abnormalities. Congenital LQTS management involves beta blockers, aggressive treatment of electrolyte imbalances, avoidance of medications that prolong the QT interval, and placement of an implantable cardioverter–defibrillator (ICD).
Last updated: Dec 15, 2025
Pathophysiology:
Types of congenital LQTS:
Variety of associated conditions:
The diagnosis of long QT syndrome can be made via an ECG ECG An electrocardiogram (ECG) is a graphic representation of the electrical activity of the heart plotted against time. Adhesive electrodes are affixed to the skin surface allowing measurement of cardiac impulses from many angles. The ECG provides 3-dimensional information about the conduction system of the heart, the myocardium, and other cardiac structures. Electrocardiogram (ECG) of the patient and/or 1st-degree relatives. A careful medication review is indicated for all patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship.
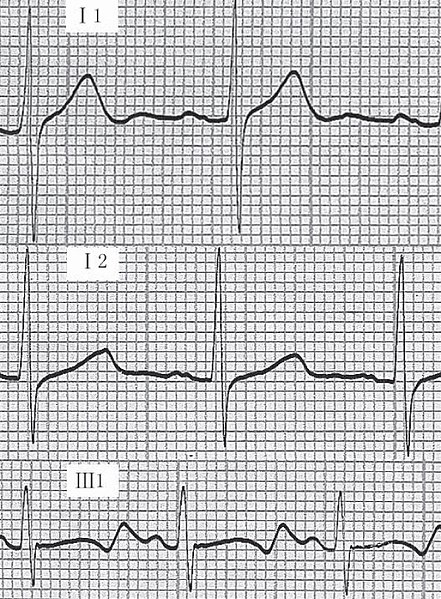
Example of an ECG tracing showing:
i) normal tracing;
ii) Romano-Ward syndrome (prolonged QT);
iii) Jervell-Lange-Nielsen syndrome (prolonged QT).
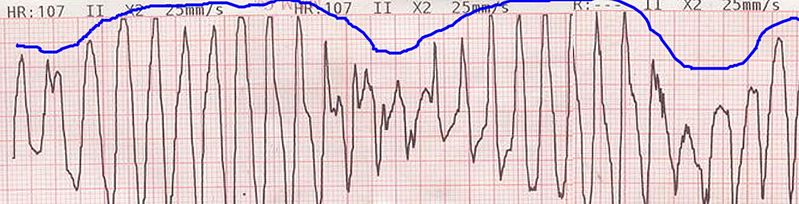
Example of an ECG tracing showing beat-to-beat axis deviation of the QRS complexes around the baseline. This is known as torsades de pointes or polymorphic ventricular tachycardia.
Image: “Tosadesdepointes” by Panthro. License: Public Domain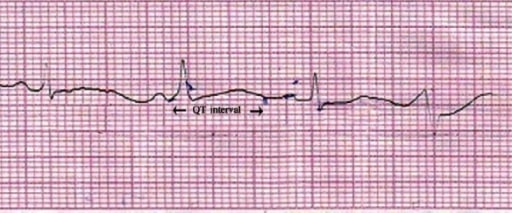
Lead II of ECG showing QT prolongation (QTc = 550 ms). Image: “Lead II of ECG showing QT prolongation” by Department of Pharmacology, Burdwan Medical College, Burdwan, West Bengal 713104, India. License: CC BY 2.0
The following medical conditions may predispose individuals to QT prolongation:
The following conditions may be included in a differential diagnosis for prolonged QT interval QT interval Electrocardiogram (ECG):
Untreated QT prolongation may evolve into the following complications: