


A síndrome do QT longo (SQTL) é um distúrbio da repolarização do miocárdio ventricular que resulta no prolongamento do QT no electrocardiograma ( ECG ECG An electrocardiogram (ECG) is a graphic representation of the electrical activity of the heart plotted against time. Adhesive electrodes are affixed to the skin surface allowing measurement of cardiac impulses from many angles. The ECG provides 3-dimensional information about the conduction system of the heart, the myocardium, and other cardiac structures. Electrocardiogram (ECG)). A síndrome do QT longo está associada a um risco aumentado de desenvolvimento de arritmias cardíacas com risco de vida, nomeadamente a torsades de pointes Torsades de pointes A malignant form of polymorphic ventricular tachycardia that is characterized by heart rate between 200 and 250 beats per minute, and QRS complexes with changing amplitude and twisting of the points. The term also describes the syndrome of tachycardia with prolonged ventricular repolarization, long qt intervals exceeding 500 milliseconds or bradycardia. Torsades de pointes may be self-limited or may progress to ventricular fibrillation. Ventricular Tachycardia. Esta patologia pode ser congénita ou adquirida. A SQTL congénita é causada por mutações genéticas que afetam os canais iónicos cardíacos. A SQTL adquirida resulta geralmente de terapêutica farmacológica ou de anomalias eletrolíticas. Os doentes podem ser assintomáticos ou apresentar palpitações, síncope, convulsões out até morte cardíaca súbita. O diagnóstico é estabelecido com um ECG ECG An electrocardiogram (ECG) is a graphic representation of the electrical activity of the heart plotted against time. Adhesive electrodes are affixed to the skin surface allowing measurement of cardiac impulses from many angles. The ECG provides 3-dimensional information about the conduction system of the heart, the myocardium, and other cardiac structures. Electrocardiogram (ECG) juntamente com a história médica e familiar, exames laboratoriais e outros testes Testes Gonadal Hormones cardíacos. O tratamento é determinado pela etiologia. A SQTL adquirida requer a suspensão do fármaco ou combinações de fármacos ofensivos e a correção das alterações eletrolíticas. O tratamento da SQTL congénita envolve bloqueadores beta, tratamento agressivo dos desequilíbrios eletrolíticos, evicção de medicamentos que prolongam o intervalo QT e colocação de um cardioversor-desfibrilhador implantável (CDI).
Last updated: Dec 15, 2025
Fisiopatologia:
Tipos de SQTL congénito:
Variedade de doenças associadas:
O diagnóstico de síndrome do QT longo pode ser feito através de um ECG ECG An electrocardiogram (ECG) is a graphic representation of the electrical activity of the heart plotted against time. Adhesive electrodes are affixed to the skin surface allowing measurement of cardiac impulses from many angles. The ECG provides 3-dimensional information about the conduction system of the heart, the myocardium, and other cardiac structures. Electrocardiogram (ECG) do doente e/ou dos familiares de 1º grau.
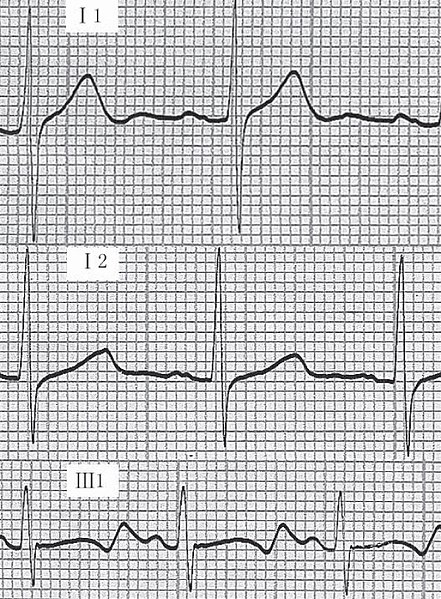
Exemplo de um traçado de ECG que mostra:
i) traçado normal;
ii) síndrome de Romano-Ward (QT prolongado);
iii) síndrome de Jervell-Lange-Nielsen (QT prolongado).
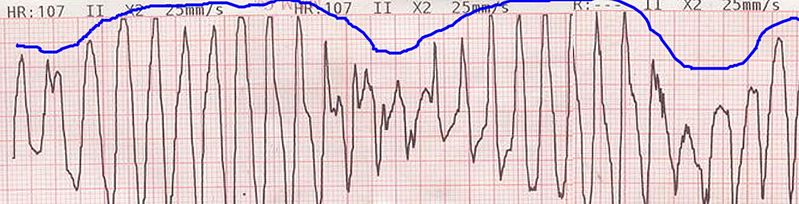
Exemplo de um traçado de ECG que mostra o desvio do eixo, batimento a batimento, dos complexos QRS em torno da linha de base. Isto é conhecido como torsades de pointes ou taquicardia ventricular polimórfica.
Imagem: “Tosadesdepointes” de Panthro. Licença: Domínio Público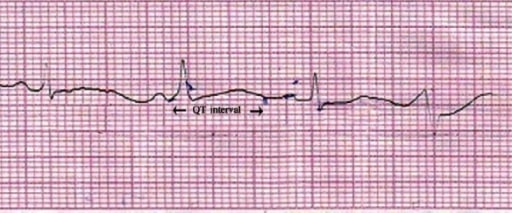
Derivação II do ECG com prolongamento do QT (QTc = 550 ms) . Imagem: “Lead II of ECG showing QT prolongation” do Department of Pharmacology, Burdwan Medical College, Burdwan, West Bengal 713104, India. Licença: CC BY 2.0
As seguintes condições médicas podem predispor ao prolongamento do intervalo QT:
As seguintes patologias podem estar incluídas no diagnóstico diferencial de intervalo QT prolongado:
O prolongamento do intervalo QT não tratado pode evoluir para as seguintes complicações:
