El síndrome de QT largo es un trastorno de la repolarización miocárdica ventricular que produce una prolongación del QT en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el electrocardiograma ( ECG ECG An electrocardiogram (ECG) is a graphic representation of the electrical activity of the heart plotted against time. Adhesive electrodes are affixed to the skin surface allowing measurement of cardiac impulses from many angles. The ECG provides 3-dimensional information about the conduction system of the heart, the myocardium, and other cardiac structures. Electrocardiogram (ECG)). El síndrome de QT largo se asocia a un mayor riesgo de desarrollar arritmias cardiacas potencialmente mortales, en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum concreto torsades de pointes Torsades de pointes A malignant form of polymorphic ventricular tachycardia that is characterized by heart rate between 200 and 250 beats per minute, and QRS complexes with changing amplitude and twisting of the points. The term also describes the syndrome of tachycardia with prolonged ventricular repolarization, long qt intervals exceeding 500 milliseconds or bradycardia. Torsades de pointes may be self-limited or may progress to ventricular fibrillation. Ventricular Tachycardia. La enfermedad puede ser congénita o adquirida. El SQTL congénito se atribuye a mutaciones genéticas que afectan a los LOS Neisseria canales iónicos cardíacos. El SQTL adquirido suele ser consecuencia de un tratamiento farmacológico o de anomalías electrolíticas. Los LOS Neisseria pacientes pueden ser asintomáticos o presentar palpitaciones, síncope, convulsiones e incluso muerte súbita cardiaca. El diagnóstico se establece con un ECG ECG An electrocardiogram (ECG) is a graphic representation of the electrical activity of the heart plotted against time. Adhesive electrodes are affixed to the skin surface allowing measurement of cardiac impulses from many angles. The ECG provides 3-dimensional information about the conduction system of the heart, the myocardium, and other cardiac structures. Electrocardiogram (ECG) junto con los LOS Neisseria antecedentes médicos y familiares, pruebas de laboratorio y otras pruebas cardiacas. El tratamiento depende de la etiología. El síndrome de QT largo adquirido requiere la retirada del medicamento o combinaciones de medicamentos causantes y la corrección de las anomalías electrolíticas. El tratamiento del síndrome del QT largo congénito incluye betabloqueantes, un tratamiento agresivo de los LOS Neisseria desequilibrios electrolíticos, la evitación de medicamentos que prolonguen el intervalo QT y la colocación de un desfibrilador cardioversor implantable.
Last updated: Dec 15, 2025
Fisiopatología:
Tipos de síndrome del QT largo congénito:
Variedad de condiciones asociadas:
El diagnóstico del síndrome del QT largo puede realizarse mediante un ECG ECG An electrocardiogram (ECG) is a graphic representation of the electrical activity of the heart plotted against time. Adhesive electrodes are affixed to the skin surface allowing measurement of cardiac impulses from many angles. The ECG provides 3-dimensional information about the conduction system of the heart, the myocardium, and other cardiac structures. Electrocardiogram (ECG) del paciente y/o de sus familiares de 1er grado.
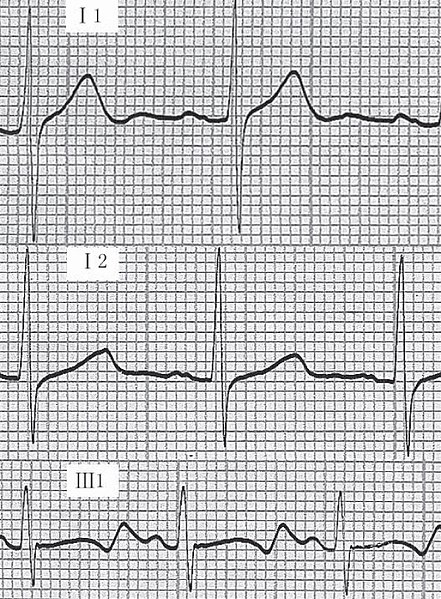
Ejemplo de un trazado de ECG que muestra:
i) trazado normal;
ii) Síndrome de Romano-Ward (QT prolongado);
iii) Síndrome de Jervell-Lange-Nielsen (QT prolongado).
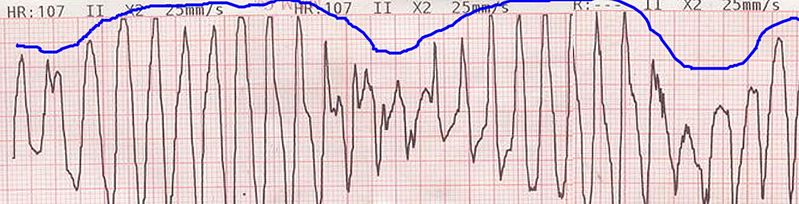
Ejemplo de un trazado de ECG que muestra la desviación del eje de los complejos QRS alrededor de la línea de base. Esto se conoce como torsades de pointes o taquicardia ventricular polimórfica.
Imagen: “Tosadesdepointes” por Panthro. Licencia: Dominio Público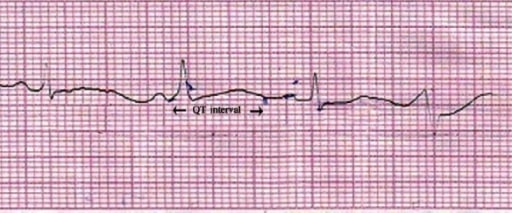
La derivada II del ECG muestra una prolongación del QT (QTc = 550 ms). Imagen: “Lead II of ECG showing QT prolongation” por Department of Pharmacology, Burdwan Medical College, Burdwan, West Bengal 713104, India. Licencia: CC BY 2.0
Las siguientes condiciones médicas pueden predisponer comúnmente a la prolongación del QT:
Las siguientes condiciones pueden incluirse en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el diagnóstico diferencial del intervalo QT prolongado:
La prolongación del QT no tratada puede evolucionar hacia las siguientes complicaciones: