Gitelman syndrome is a rare genetic autosomal recessive disorder that affects the sodium-chloride cotransporter in the distal convoluted tubule of the nephron and causes electrolyte abnormalities. The syndrome presents clinically with symptoms of hypokalemia and hypomagnesemia. Diagnosis is based on clinical presentation and laboratory testing (showing hypokalemia, hypomagnesemia, metabolic alkalosis, and hypocalciuria), and confirmed with genetic testing. The mainstay of management is electrolyte supplementation. The prognosisPrognosisA prediction of the probable outcome of a disease based on a individual's condition and the usual course of the disease as seen in similar situations.Non-Hodgkin Lymphomas is good, but hypokalemiaHypokalemiaHypokalemia is defined as plasma potassium (K+) concentration < 3.5 mEq/L. Homeostatic mechanisms maintain plasma concentration between 3.5-5.2 mEq/L despite marked variation in dietary intake. Hypokalemia can be due to renal losses, GI losses, transcellular shifts, or poor dietary intake.Hypokalemia must be monitored to prevent cardiac arrhythmias and potential cardiac arrestCardiac arrestCardiac arrest is the sudden, complete cessation of cardiac output with hemodynamic collapse. Patients present as pulseless, unresponsive, and apneic. Rhythms associated with cardiac arrest are ventricular fibrillation/tachycardia, asystole, or pulseless electrical activity. Cardiac Arrest.
Gitelman syndrome (GS) is a rare genetic autosomal recessiveAutosomal recessiveAutosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal recessive diseases are only expressed when 2 copies of the recessive allele are inherited.Autosomal Recessive and Autosomal Dominant Inheritance disorder that affects the thiazide-sensitive Na+–Cl– cotransporter (NCC) in the distal convoluted tubule (DCT), leading to a salt-wasting tubulopathy.
Epidemiology
PrevalencePrevalenceThe total number of cases of a given disease in a specified population at a designated time. It is differentiated from incidence, which refers to the number of new cases in the population at a given time.Measures of Disease Frequency:
1–10 per 40,000 individuals
Higher prevalencePrevalenceThe total number of cases of a given disease in a specified population at a designated time. It is differentiated from incidence, which refers to the number of new cases in the population at a given time.Measures of Disease Frequency in East Asian populations, estimated at 2.3 per 1,000 individuals
More common than Bartter syndromeBartter syndromeBartter syndrome is a rare autosomal recessive disorder that affects the kidneys and presents either antenatally with severe or life-threatening manifestations or in childhood or adulthood with a milder course, depending on the genetic defect. Clinical disease results from defective renal reabsorption of sodium chloride in the thick ascending limb of the loop of Henle.Bartter Syndrome
No sexSexThe totality of characteristics of reproductive structure, functions, phenotype, and genotype, differentiating the male from the female organism.Gender Dysphoria predisposition
Etiology
Autosomal recessive inheritanceAutosomal recessive inheritanceAutosomal Recessive and Autosomal Dominant Inheritance: For a person to be affected, a mutationMutationGenetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations must be present on both genesGenesA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.DNA Types and Structure encoding the NCC.
CarrierCarrierVaccination: A person with only 1 mutated geneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics remains unaffected.
A 50% chance of the offspring being an unaffected carrierCarrierVaccination
A 25% chance of the offspring being unaffected and not a carrierCarrierVaccination
The mutationMutationGenetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations is present in the SLC12A3geneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics (80% of individuals) and, rarely, in the CLCNKBgeneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics (which is also implicated in Bartter syndromeBartter syndromeBartter syndrome is a rare autosomal recessive disorder that affects the kidneys and presents either antenatally with severe or life-threatening manifestations or in childhood or adulthood with a milder course, depending on the genetic defect. Clinical disease results from defective renal reabsorption of sodium chloride in the thick ascending limb of the loop of Henle.Bartter Syndrome type III).
The DCT is the smallest portion of the duct system in a nephronNephronThe functional units of the kidney, consisting of the glomerulus and the attached tubule.Kidneys: Anatomy. It measures about 5 mm in size and starts from the maculaMaculaAn oval area in the retina, 3 to 5 mm in diameter, usually located temporal to the posterior pole of the eye and slightly below the level of the optic disk. It is characterized by the presence of a yellow pigment diffusely permeating the inner layers, contains the fovea centralis in its center, and provides the best phototropic visual acuity. It is devoid of retinal blood vessels, except in its periphery, and receives nourishment from the choriocapillaris of the choroid.Eye: Anatomy densa. Characteristics of the DCT are as follows:
Presence of a large number of mitochondriaMitochondriaSemiautonomous, self-reproducing organelles that occur in the cytoplasm of all cells of most, but not all, eukaryotes. Each mitochondrion is surrounded by a double limiting membrane. The inner membrane is highly invaginated, and its projections are called cristae. Mitochondria are the sites of the reactions of oxidative phosphorylation, which result in the formation of ATP. They contain distinctive ribosomes, transfer RNAs; amino Acyl tRNA synthetases; and elongation and termination factors. Mitochondria depend upon genes within the nucleus of the cells in which they reside for many essential messenger RNAs. Mitochondria are believed to have arisen from aerobic bacteria that established a symbiotic relationship with primitive protoeukaryotes.The Cell: Organelles and Na+/K+-ATPase pumps in the basolateral membrane
Impermeable to water and ureaUreaA compound formed in the liver from ammonia produced by the deamination of amino acids. It is the principal end product of protein catabolism and constitutes about one half of the total urinary solids.Urea Cycle
Contains the NCC, which is the target of thiazideThiazideHeterocyclic compounds with sulfur and nitrogen in the ring. This term commonly refers to the benzothiadiazines that inhibit sodium-potassium-chloride symporters and are used as diuretics.HyponatremiadiureticsDiureticsAgents that promote the excretion of urine through their effects on kidney function.Heart Failure and Chronic Coronary Syndrome Medication
Functions of the DCT:
Regulation of pHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance by secretionSecretionCoagulation Studies of H+ ions and reabsorption of HCO3–, or vice versa
Reabsorption of Na+, Cl–, Mg2+, and calciumCalciumA basic element found in nearly all tissues. It is a member of the alkaline earth family of metals with the atomic symbol ca, atomic number 20, and atomic weight 40. Calcium is the most abundant mineral in the body and combines with phosphorus to form calcium phosphate in the bones and teeth. It is essential for the normal functioning of nerves and muscles and plays a role in blood coagulation (as factor IV) and in many enzymatic processes.Electrolytes (CaCACondylomata acuminata are a clinical manifestation of genital HPV infection. Condylomata acuminata are described as raised, pearly, flesh-colored, papular, cauliflower-like lesions seen in the anogenital region that may cause itching, pain, or bleeding.Condylomata Acuminata (Genital Warts)2+)
Gitelman syndrome leads to the loss of functionLoss of FunctionInflammation of the NCC → electrolyte abnormalities due to interference with the normal functioning of the DCT
GeneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics mutations inactivate the NCC:
Normally functions to reabsorb Na+ and Cl– from the DCT
Inactivation prevents reabsorption → ↑ Na+ and Cl– delivery to the collecting ductCollecting ductStraight tubes commencing in the radiate part of the kidney cortex where they receive the curved ends of the distal convoluted tubules. In the medulla the collecting tubules of each pyramid converge to join a central tube (duct of bellini) which opens on the summit of the papilla.Renal Cell Carcinoma
Impaired salt reabsorption → ↑ water loss
Volume contraction → activation of RAASRAASA blood pressure regulating system of interacting components that include renin; angiotensinogen; angiotensin converting enzyme; angiotensin i; angiotensin ii; and angiotensinase. Renin, an enzyme produced in the kidney, acts on angiotensinogen, an alpha-2 globulin produced by the liver, forming angiotensin I. Angiotensin-converting enzyme, contained in the lung, acts on angiotensin I in the plasma converting it to angiotensin II, an extremely powerful vasoconstrictor. Angiotensin II causes contraction of the arteriolar and renal vascular smooth muscle, leading to retention of salt and water in the kidney and increased arterial blood pressure. In addition, angiotensin II stimulates the release of aldosterone from the adrenal cortex, which in turn also increases salt and water retention in the kidney. Angiotensin-converting enzyme also breaks down bradykinin, a powerful vasodilator and component of the kallikrein-kinin system.Adrenal Hormones (even with RAASRAASA blood pressure regulating system of interacting components that include renin; angiotensinogen; angiotensin converting enzyme; angiotensin i; angiotensin ii; and angiotensinase. Renin, an enzyme produced in the kidney, acts on angiotensinogen, an alpha-2 globulin produced by the liver, forming angiotensin I. Angiotensin-converting enzyme, contained in the lung, acts on angiotensin I in the plasma converting it to angiotensin II, an extremely powerful vasoconstrictor. Angiotensin II causes contraction of the arteriolar and renal vascular smooth muscle, leading to retention of salt and water in the kidney and increased arterial blood pressure. In addition, angiotensin II stimulates the release of aldosterone from the adrenal cortex, which in turn also increases salt and water retention in the kidney. Angiotensin-converting enzyme also breaks down bradykinin, a powerful vasodilator and component of the kallikrein-kinin system.Adrenal Hormones activation, typically normal or low blood pressure is noted, possibly due to the chronic nature of the volume depletionVolume depletionVolume status is a balance between water and solutes, the majority of which is Na. Volume depletion refers to a loss of both water and Na, whereas dehydration refers only to a loss of water. Volume depletion can be caused by GI losses, renal losses, bleeding, poor oral Na intake, or third spacing of fluids.Volume Depletion and Dehydration and compensatory mechanisms)
↑ ReninReninA highly specific (leu-leu) endopeptidase that generates angiotensin I from its precursor angiotensinogen, leading to a cascade of reactions which elevate blood pressure and increase sodium retention by the kidney in the renin-angiotensin system.Renal Sodium and Water Regulation and aldosteroneAldosteroneA hormone secreted by the adrenal cortex that regulates electrolyte and water balance by increasing the renal retention of sodium and the excretion of potassium.Hyperkalemia activity in the collecting ductCollecting ductStraight tubes commencing in the radiate part of the kidney cortex where they receive the curved ends of the distal convoluted tubules. In the medulla the collecting tubules of each pyramid converge to join a central tube (duct of bellini) which opens on the summit of the papilla.Renal Cell Carcinoma results in:
↑ K+ excretion → hypokalemiaHypokalemiaHypokalemia is defined as plasma potassium (K+) concentration < 3.5 mEq/L. Homeostatic mechanisms maintain plasma concentration between 3.5-5.2 mEq/L despite marked variation in dietary intake. Hypokalemia can be due to renal losses, GI losses, transcellular shifts, or poor dietary intake.Hypokalemia
↑ H+ excretion → metabolic alkalosisAlkalosisA pathological condition that removes acid or adds base to the body fluids.Respiratory Alkalosis
Additionally:
↓ Mg2+ reabsorption → renal Mg2+ wasting → hypomagnesemiaHypomagnesemiaA nutritional condition produced by a deficiency of magnesium in the diet, characterized by anorexia, nausea, vomiting, lethargy, and weakness. Symptoms are paresthesias, muscle cramps, irritability, decreased attention span, and mental confusion, possibly requiring months to appear. Deficiency of body magnesium can exist even when serum values are normal. In addition, magnesium deficiency may be organ-selective, since certain tissues become deficient before others. Electrolytes
↓ Urinary calciumCalciumA basic element found in nearly all tissues. It is a member of the alkaline earth family of metals with the atomic symbol ca, atomic number 20, and atomic weight 40. Calcium is the most abundant mineral in the body and combines with phosphorus to form calcium phosphate in the bones and teeth. It is essential for the normal functioning of nerves and muscles and plays a role in blood coagulation (as factor IV) and in many enzymatic processes.Electrolytes excretion
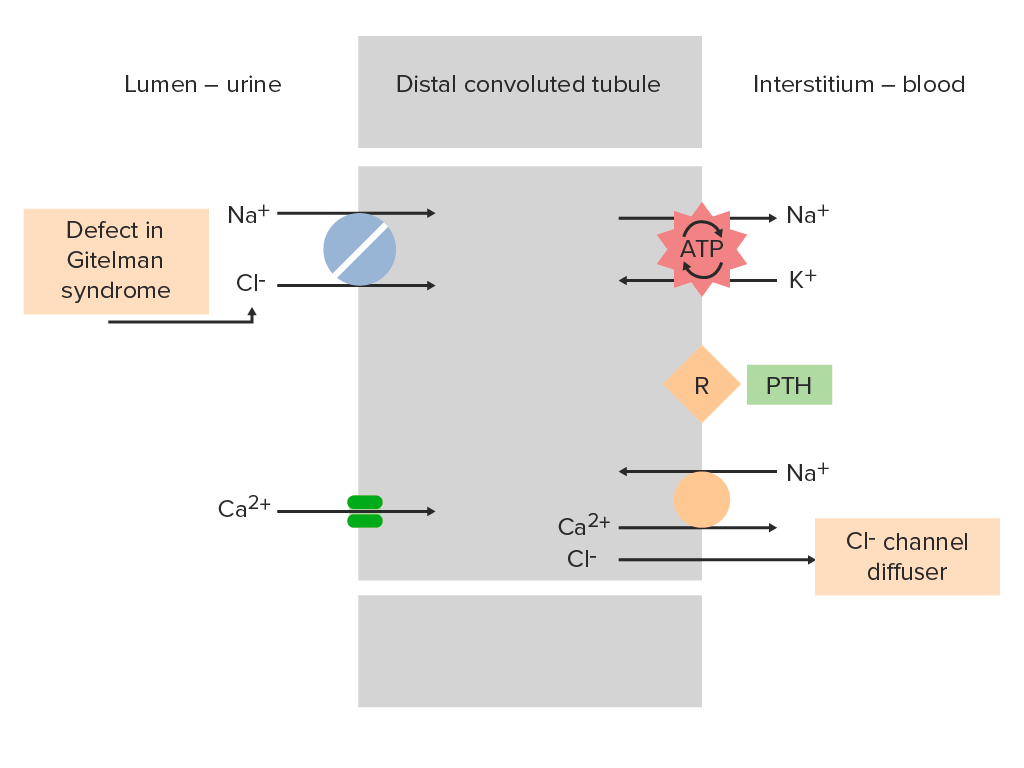
Sodium-chloride cotransporter (NCC) in the distal convoluted tubule (DCT) of a nephron:
NCC normally helps with reabsorption of Na+ and Cl- from the tubule lumen. In Gitelman syndrome, inactivation of the NCC prevents this absorption, increasing electrolyte delivery to the collecting duct. This phenomenon is similar to the mechanism of action of thiazide diuretics.
PTH: parathyroid hormone
R: parathyroid hormone receptor
Image by Lecturio.
Anatomy of a nephron:
The green represents the distal convoluted tubule, which is affected in individuals with Gitelman syndrome.
Image by Lecturio.
Clinical Presentation
Individuals with GS have mild-to-moderate symptoms without limitation in daily activity. These individuals present after the 1st decade of life in adolescence or early adulthood (rarely in infancy).
The classic clinical presentation is the triad of:
HypokalemiaHypokalemiaHypokalemia is defined as plasma potassium (K+) concentration < 3.5 mEq/L. Homeostatic mechanisms maintain plasma concentration between 3.5-5.2 mEq/L despite marked variation in dietary intake. Hypokalemia can be due to renal losses, GI losses, transcellular shifts, or poor dietary intake.Hypokalemia
Metabolic alkalosisAlkalosisA pathological condition that removes acid or adds base to the body fluids.Respiratory Alkalosis
Normal or low blood pressure
Other signs and symptoms include:
Manifestations of hypokalemiaHypokalemiaHypokalemia is defined as plasma potassium (K+) concentration < 3.5 mEq/L. Homeostatic mechanisms maintain plasma concentration between 3.5-5.2 mEq/L despite marked variation in dietary intake. Hypokalemia can be due to renal losses, GI losses, transcellular shifts, or poor dietary intake.Hypokalemia:
ConstipationConstipationConstipation is common and may be due to a variety of causes. Constipation is generally defined as bowel movement frequency < 3 times per week. Patients who are constipated often strain to pass hard stools. The condition is classified as primary (also known as idiopathic or functional constipation) or secondary, and as acute or chronic. Constipation
Severe fatigueFatigueThe state of weariness following a period of exertion, mental or physical, characterized by a decreased capacity for work and reduced efficiency to respond to stimuli.Fibromyalgia
Cardiac arrhythmias and cardiac arrestCardiac arrestCardiac arrest is the sudden, complete cessation of cardiac output with hemodynamic collapse. Patients present as pulseless, unresponsive, and apneic. Rhythms associated with cardiac arrest are ventricular fibrillation/tachycardia, asystole, or pulseless electrical activity. Cardiac Arrest (in severe cases)
Manifestations of hypomagnesemiaHypomagnesemiaA nutritional condition produced by a deficiency of magnesium in the diet, characterized by anorexia, nausea, vomiting, lethargy, and weakness. Symptoms are paresthesias, muscle cramps, irritability, decreased attention span, and mental confusion, possibly requiring months to appear. Deficiency of body magnesium can exist even when serum values are normal. In addition, magnesium deficiency may be organ-selective, since certain tissues become deficient before others. Electrolytes:
ParesthesiasParesthesiasSubjective cutaneous sensations (e.g., cold, warmth, tingling, pressure, etc.) that are experienced spontaneously in the absence of stimulation.Posterior Cord Syndrome, especially in the face
TetanyTetanyA disorder characterized by muscle twitches, cramps, and carpopedal spasm, and when severe, laryngospasm and seizures. This condition is associated with unstable depolarization of axonal membranes, primarily in the peripheral nervous system. Tetany usually results from hypocalcemia or reduced serum levels of magnesium that may be associated with hyperventilation; hypoparathyroidism; rickets; uremia; or other conditions.Hypocalcemia
Chondrocalcinosis in adulthood: associated with inflammationInflammationInflammation is a complex set of responses to infection and injury involving leukocytes as the principal cellular mediators in the body’s defense against pathogenic organisms. Inflammation is also seen as a response to tissue injury in the process of wound healing. The 5 cardinal signs of inflammation are pain, heat, redness, swelling, and loss of function. Inflammation of joints
SeizuresSeizuresA seizure is abnormal electrical activity of the neurons in the cerebral cortex that can manifest in numerous ways depending on the region of the brain affected. Seizures consist of a sudden imbalance that occurs between the excitatory and inhibitory signals in cortical neurons, creating a net excitation. The 2 major classes of seizures are focal and generalized. Seizures in severe cases
Growth delay is seen in individuals affected at a young age.
Diagnosis
A detailed evaluation of GS is necessary when an individual presents with unexplained hypokalemiaHypokalemiaHypokalemia is defined as plasma potassium (K+) concentration < 3.5 mEq/L. Homeostatic mechanisms maintain plasma concentration between 3.5-5.2 mEq/L despite marked variation in dietary intake. Hypokalemia can be due to renal losses, GI losses, transcellular shifts, or poor dietary intake.Hypokalemia, metabolic alkalosisAlkalosisA pathological condition that removes acid or adds base to the body fluids.Respiratory Alkalosis, and normal or low blood pressure. Because of its rare occurrence compared with other renal disorders with similar symptoms, other causes should be ruled out.
Lab tests
Serum electrolyte levels:
↓ K+
↓ or normal Mg2+
↑ bicarbonateBicarbonateInorganic salts that contain the -HCO3 radical. They are an important factor in determining the ph of the blood and the concentration of bicarbonate ions is regulated by the kidney. Levels in the blood are an index of the alkali reserve or buffering capacity.Electrolytes
↑ ReninReninA highly specific (leu-leu) endopeptidase that generates angiotensin I from its precursor angiotensinogen, leading to a cascade of reactions which elevate blood pressure and increase sodium retention by the kidney in the renin-angiotensin system.Renal Sodium and Water Regulation
↑ AldosteroneAldosteroneA hormone secreted by the adrenal cortex that regulates electrolyte and water balance by increasing the renal retention of sodium and the excretion of potassium.Hyperkalemia
↑ Blood pHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance (alkalosisAlkalosisA pathological condition that removes acid or adds base to the body fluids.Respiratory Alkalosis)
Urinary tests:
Na+: ↑ excretion
K+: ↑ excretion
Cl–: ↑ excretion
Mg2+: ↑ excretion
CaCACondylomata acuminata are a clinical manifestation of genital HPV infection. Condylomata acuminata are described as raised, pearly, flesh-colored, papular, cauliflower-like lesions seen in the anogenital region that may cause itching, pain, or bleeding.Condylomata Acuminata (Genital Warts)2+: ↓ excretion
Genetic testingGenetic TestingDetection of a mutation; genotype; karyotype; or specific alleles associated with genetic traits, heritable diseases, or predisposition to a disease, or that may lead to the disease in descendants. It includes prenatal genetic testing.Myotonic Dystrophies
Genetic testingGenetic TestingDetection of a mutation; genotype; karyotype; or specific alleles associated with genetic traits, heritable diseases, or predisposition to a disease, or that may lead to the disease in descendants. It includes prenatal genetic testing.Myotonic Dystrophies is highly specific and sensitive, and a majority of affected individuals show mutations in 2 particular genesGenesA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.DNA Types and Structure:
SLC12A3 (80% of affected individuals)
CLCNKB
Management and Prognosis
Management
The goals of management are to minimize the effects of extracellular volume depletionVolume depletionVolume status is a balance between water and solutes, the majority of which is Na. Volume depletion refers to a loss of both water and Na, whereas dehydration refers only to a loss of water. Volume depletion can be caused by GI losses, renal losses, bleeding, poor oral Na intake, or third spacing of fluids.Volume Depletion and Dehydration and correct electrolyte deficiencies.
Asymptomatic individuals:
Need regularRegularInsulin physician evaluation for electrolyte imbalances
Lab tests for electrolytesElectrolytesElectrolytes are mineral salts that dissolve in water and dissociate into charged particles called ions, which can be either be positively (cations) or negatively (anions) charged. Electrolytes are distributed in the extracellular and intracellular compartments in different concentrations. Electrolytes are essential for various basic life-sustaining functions.Electrolytes every 6–12 months
HypomagnesemiaHypomagnesemiaA nutritional condition produced by a deficiency of magnesium in the diet, characterized by anorexia, nausea, vomiting, lethargy, and weakness. Symptoms are paresthesias, muscle cramps, irritability, decreased attention span, and mental confusion, possibly requiring months to appear. Deficiency of body magnesium can exist even when serum values are normal. In addition, magnesium deficiency may be organ-selective, since certain tissues become deficient before others. Electrolytes:
Lifelong supplementation with oral magnesiumMagnesiumA metallic element that has the atomic symbol mg, atomic number 12, and atomic weight 24. 31. It is important for the activity of many enzymes, especially those involved in oxidative phosphorylation.ElectrolyteschlorideChlorideInorganic compounds derived from hydrochloric acid that contain the Cl- ion.Electrolytes in 3–4 divided doses/day
TetanyTetanyA disorder characterized by muscle twitches, cramps, and carpopedal spasm, and when severe, laryngospasm and seizures. This condition is associated with unstable depolarization of axonal membranes, primarily in the peripheral nervous system. Tetany usually results from hypocalcemia or reduced serum levels of magnesium that may be associated with hyperventilation; hypoparathyroidism; rickets; uremia; or other conditions.Hypocalcemia requires IV magnesiumMagnesiumA metallic element that has the atomic symbol mg, atomic number 12, and atomic weight 24. 31. It is important for the activity of many enzymes, especially those involved in oxidative phosphorylation.ElectrolyteschlorideChlorideInorganic compounds derived from hydrochloric acid that contain the Cl- ion.Electrolytes.
Note that correcting magnesium deficiencyMagnesium DeficiencyA nutritional condition produced by a deficiency of magnesium in the diet, characterized by anorexia, nausea, vomiting, lethargy, and weakness. Symptoms are paresthesias, muscle cramps, irritability, decreased attention span, and mental confusion, possibly requiring months to appear. Deficiency of body magnesium can exist even when serum values are normal. In addition, magnesium deficiency may be organ-selective, since certain tissues become deficient before others.Electrolytes is essential for effective potassiumPotassiumAn element in the alkali group of metals with an atomic symbol k, atomic number 19, and atomic weight 39. 10. It is the chief cation in the intracellular fluid of muscle and other cells. Potassium ion is a strong electrolyte that plays a significant role in the regulation of fluid volume and maintenance of the water-electrolyte balance.Hyperkalemia repletion. MagnesiumMagnesiumA metallic element that has the atomic symbol mg, atomic number 12, and atomic weight 24. 31. It is important for the activity of many enzymes, especially those involved in oxidative phosphorylation.Electrolytes replacement is initiated first (before potassiumPotassiumAn element in the alkali group of metals with an atomic symbol k, atomic number 19, and atomic weight 39. 10. It is the chief cation in the intracellular fluid of muscle and other cells. Potassium ion is a strong electrolyte that plays a significant role in the regulation of fluid volume and maintenance of the water-electrolyte balance.Hyperkalemia).
HypokalemiaHypokalemiaHypokalemia is defined as plasma potassium (K+) concentration < 3.5 mEq/L. Homeostatic mechanisms maintain plasma concentration between 3.5-5.2 mEq/L despite marked variation in dietary intake. Hypokalemia can be due to renal losses, GI losses, transcellular shifts, or poor dietary intake.Hypokalemia:
K+ levels should be monitored to avoid cardiac arrhythmias.
Potassium chloridePotassium chlorideA white crystal or crystalline powder used in buffers; fertilizers; and explosives. It can be used to replenish electrolytes and restore water-electrolyte balance in treating hypokalemia.Esophagitis supplementation will likely be needed.
Potassium-sparing diuretic:
AmilorideAmilorideA pyrazine compound inhibiting sodium reabsorption through sodium channels in renal epithelial cells. This inhibition creates a negative potential in the luminal membranes of principal cells, located in the distal convoluted tubule and collecting duct. Negative potential reduces secretion of potassium and hydrogen ions. Amiloride is used in conjunction with diuretics to spare potassium loss.Liddle Syndrome
With aldosteroneAldosteroneA hormone secreted by the adrenal cortex that regulates electrolyte and water balance by increasing the renal retention of sodium and the excretion of potassium.Hyperkalemia antagonist activity: spironolactoneSpironolactoneA potassium sparing diuretic that acts by antagonism of aldosterone in the distal renal tubules. It is used mainly in the treatment of refractory edema in patients with congestive heart failure, nephrotic syndrome, or hepatic cirrhosis. Its effects on the endocrine system are utilized in the treatments of hirsutism and acne but they can lead to adverse effects.Potassium-sparing Diuretics and eplerenoneEplerenoneA spironolactone derivative and selective aldosterone receptor antagonist that is used in the management of hypertension and congestive heart failure, post-myocardial infarction.Potassium-sparing Diuretics
Affected individuals should be carefully monitored to avoid hypotensionHypotensionHypotension is defined as low blood pressure, specifically < 90/60 mm Hg, and is most commonly a physiologic response. Hypotension may be mild, serious, or life threatening, depending on the cause. Hypotension.
ARBsARBsAgents that antagonize angiotensin receptors. Many drugs in this class specifically target the angiotensin type 1 receptor.Heart Failure and Chronic Coronary Syndrome Medication or ACEisACEIsA class of drugs whose main indications are the treatment of hypertension and heart failure. They exert their hemodynamic effect mainly by inhibiting the renin-angiotensin system. They also modulate sympathetic nervous system activity and increase prostaglandin synthesis. They cause mainly vasodilation and mild natriuresis without affecting heart rate and contractility.Heart Failure and Chronic Coronary Syndrome Medication may also be used.
SodiumSodiumA member of the alkali group of metals. It has the atomic symbol na, atomic number 11, and atomic weight 23.Hyponatremia loss:
Salt intake is encouraged.
SodiumSodiumA member of the alkali group of metals. It has the atomic symbol na, atomic number 11, and atomic weight 23.HyponatremiachlorideChlorideInorganic compounds derived from hydrochloric acid that contain the Cl- ion.Electrolytes supplementation may be needed.
Rare, for individuals with end-stage renal disease
Tubular abnormalities resolve without recurrence after kidney transplantationKidney TransplantationThe transference of a kidney from one human or animal to another.Organ Transplantation.
PrognosisPrognosisA prediction of the probable outcome of a disease based on a individual’s condition and the usual course of the disease as seen in similar situations.Non-Hodgkin Lymphomas
Generally good
The ability to perform daily life activities varies among affected individuals.
Progression to renal insufficiency is extremely rare.
GS does not affect life expectancyLife expectancyBased on known statistical data, the number of years which any person of a given age may reasonably expected to live.Population Pyramids.
Differential Diagnosis
Bartter syndromeBartter syndromeBartter syndrome is a rare autosomal recessive disorder that affects the kidneys and presents either antenatally with severe or life-threatening manifestations or in childhood or adulthood with a milder course, depending on the genetic defect. Clinical disease results from defective renal reabsorption of sodium chloride in the thick ascending limb of the loop of Henle.Bartter Syndrome: a rare genetic disorder that results in impaired NaCl reabsorption in the thick ascending limbThick ascending limbRenal Sodium and Water Regulation of the loop of HenleLoop of HenleThe U-shaped portion of the renal tubule in the kidney medulla, consisting of a descending limb and an ascending limb. It is situated between the proximal kidney tubule and the distal kidney tubule.Tubular System. Bartter syndromeBartter syndromeBartter syndrome is a rare autosomal recessive disorder that affects the kidneys and presents either antenatally with severe or life-threatening manifestations or in childhood or adulthood with a milder course, depending on the genetic defect. Clinical disease results from defective renal reabsorption of sodium chloride in the thick ascending limb of the loop of Henle.Bartter Syndrome presents with hypokalemiaHypokalemiaHypokalemia is defined as plasma potassium (K+) concentration < 3.5 mEq/L. Homeostatic mechanisms maintain plasma concentration between 3.5-5.2 mEq/L despite marked variation in dietary intake. Hypokalemia can be due to renal losses, GI losses, transcellular shifts, or poor dietary intake.Hypokalemia, metabolic alkalosisAlkalosisA pathological condition that removes acid or adds base to the body fluids.Respiratory Alkalosis, and delayed growth/development, and elevated urine calciumCalciumA basic element found in nearly all tissues. It is a member of the alkaline earth family of metals with the atomic symbol ca, atomic number 20, and atomic weight 40. Calcium is the most abundant mineral in the body and combines with phosphorus to form calcium phosphate in the bones and teeth. It is essential for the normal functioning of nerves and muscles and plays a role in blood coagulation (as factor IV) and in many enzymatic processes.Electrolytes levels. Diagnosis is by blood and urine testing. Management focuses on treating symptoms and replenishing electrolytesElectrolytesElectrolytes are mineral salts that dissolve in water and dissociate into charged particles called ions, which can be either be positively (cations) or negatively (anions) charged. Electrolytes are distributed in the extracellular and intracellular compartments in different concentrations. Electrolytes are essential for various basic life-sustaining functions.Electrolytes.
LaxativeLaxativeAgents that produce a soft formed stool, and relax and loosen the bowels, typically used over a protracted period, to relieve constipation.Hypokalemia abuse: seen in individuals with eating disorders for weight lossWeight lossDecrease in existing body weight.Bariatric Surgery. The 2 main disorders are bulimiaBulimiaEating an excess amount of food in a short period of time, as seen in the disorder of bulimia nervosa. It is caused by an abnormal craving for food, or insatiable hunger also known as ‘ox hunger’.Bulimia Nervosa and anorexiaAnorexiaThe lack or loss of appetite accompanied by an aversion to food and the inability to eat. It is the defining characteristic of the disorder anorexia nervosa.Anorexia Nervosa nervosa. The associated risks are dehydrationDehydrationThe condition that results from excessive loss of water from a living organism.Volume Depletion and Dehydration, electrolyte abnormalities, constipationConstipationConstipation is common and may be due to a variety of causes. Constipation is generally defined as bowel movement frequency < 3 times per week. Patients who are constipated often strain to pass hard stools. The condition is classified as primary (also known as idiopathic or functional constipation) or secondary, and as acute or chronic. Constipation, infectionsInfectionsInvasion of the host organism by microorganisms or their toxins or by parasites that can cause pathological conditions or diseases.Chronic Granulomatous Disease, and rectal prolapseRectal prolapseRectal prolapse, also known as rectal procidentia, is the protrusion of rectal tissue through the anus. The tissue may include just the mucosa or the full thickness of the rectal wall. Common risk factors include chronic straining, constipation, bowel motility disorders, and weakening of the pelvic floor muscles. Rectal Prolapse. It is important to treat the underlying condition and start CBT.
Diuretic abuse: another disorder wherein people attempt to lose weight. Affected individuals take excess diureticsDiureticsAgents that promote the excretion of urine through their effects on kidney function.Heart Failure and Chronic Coronary Syndrome Medication to feel lighter, which leads to dehydrationDehydrationThe condition that results from excessive loss of water from a living organism.Volume Depletion and Dehydration and electrolyte abnormalities. Diuretic abuse is approached with CBT and educating the individual about the complications of diuretic use.
Mineralocorticoid excessMineralocorticoid excessA hereditary disease characterized by childhood onset hypertension, hypokalemic alkalosis, and low renin and aldosterone secretion. It results from a defect in the activity of the 11-beta-hydroxysteroid dehydrogenase type 2 enzyme which results in inadequate conversion of cortisol to cortisone. The build up of unprocessed cortisol to levels that stimulate mineralocorticoid receptors creates the appearance of having excessive mineralocorticoids.Metabolic Alkalosis: an autosomal recessiveAutosomal recessiveAutosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal recessive diseases are only expressed when 2 copies of the recessive allele are inherited.Autosomal Recessive and Autosomal Dominant Inheritance disorder that results from mutations in the HSD11B2geneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics that encodes the kidney isozyme 11β-hydroxysteroid dehydrogenase type 2. Mineralocorticoid excessMineralocorticoid excessA hereditary disease characterized by childhood onset hypertension, hypokalemic alkalosis, and low renin and aldosterone secretion. It results from a defect in the activity of the 11-beta-hydroxysteroid dehydrogenase type 2 enzyme which results in inadequate conversion of cortisol to cortisone. The build up of unprocessed cortisol to levels that stimulate mineralocorticoid receptors creates the appearance of having excessive mineralocorticoids.Metabolic Alkalosis presents with hypertensionHypertensionHypertension, or high blood pressure, is a common disease that manifests as elevated systemic arterial pressures. Hypertension is most often asymptomatic and is found incidentally as part of a routine physical examination or during triage for an unrelated medical encounter. Hypertension, hypokalemiaHypokalemiaHypokalemia is defined as plasma potassium (K+) concentration < 3.5 mEq/L. Homeostatic mechanisms maintain plasma concentration between 3.5-5.2 mEq/L despite marked variation in dietary intake. Hypokalemia can be due to renal losses, GI losses, transcellular shifts, or poor dietary intake.Hypokalemia, metabolic alkalosisAlkalosisA pathological condition that removes acid or adds base to the body fluids.Respiratory Alkalosis, and low plasmaPlasmaThe residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation.Transfusion ProductsreninReninA highly specific (leu-leu) endopeptidase that generates angiotensin I from its precursor angiotensinogen, leading to a cascade of reactions which elevate blood pressure and increase sodium retention by the kidney in the renin-angiotensin system.Renal Sodium and Water Regulation. Diagnosis is made on finding the triad of hypertensionHypertensionHypertension, or high blood pressure, is a common disease that manifests as elevated systemic arterial pressures. Hypertension is most often asymptomatic and is found incidentally as part of a routine physical examination or during triage for an unrelated medical encounter. Hypertension, hypokalemiaHypokalemiaHypokalemia is defined as plasma potassium (K+) concentration < 3.5 mEq/L. Homeostatic mechanisms maintain plasma concentration between 3.5-5.2 mEq/L despite marked variation in dietary intake. Hypokalemia can be due to renal losses, GI losses, transcellular shifts, or poor dietary intake.Hypokalemia, and suppressed plasmaPlasmaThe residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation.Transfusion ProductsaldosteroneAldosteroneA hormone secreted by the adrenal cortex that regulates electrolyte and water balance by increasing the renal retention of sodium and the excretion of potassium.Hyperkalemia levels plus an abnormal urinary cortisol-to-cortisone ratio. Management includes aldosterone antagonistsAldosterone antagonistsDrugs that bind to and block the activation of mineralocorticoid receptors by mineralocorticoids such as aldosterone.Heart Failure and Chronic Coronary Syndrome Medication such as spironolactoneSpironolactoneA potassium sparing diuretic that acts by antagonism of aldosterone in the distal renal tubules. It is used mainly in the treatment of refractory edema in patients with congestive heart failure, nephrotic syndrome, or hepatic cirrhosis. Its effects on the endocrine system are utilized in the treatments of hirsutism and acne but they can lead to adverse effects.Potassium-sparing Diuretics.