Bartter syndrome is a rare autosomal recessiveAutosomal recessiveAutosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal recessive diseases are only expressed when 2 copies of the recessive allele are inherited.Autosomal Recessive and Autosomal Dominant Inheritance disorder that affects the kidneysKidneysThe kidneys are a pair of bean-shaped organs located retroperitoneally against the posterior wall of the abdomen on either side of the spine. As part of the urinary tract, the kidneys are responsible for blood filtration and excretion of water-soluble waste in the urine.Kidneys: Anatomy and presents either antenatally with severe or life-threatening manifestations or in childhood or adulthood with a milder course, depending on the genetic defectGenetic DefectIon Channel Myopathy. Clinical disease results from defective renal reabsorption of sodiumSodiumA member of the alkali group of metals. It has the atomic symbol na, atomic number 11, and atomic weight 23.HyponatremiachlorideChlorideInorganic compounds derived from hydrochloric acid that contain the Cl- ion.Electrolytes in the thick ascending limbThick ascending limbRenal Sodium and Water Regulation of the loop of HenleLoop of HenleThe U-shaped portion of the renal tubule in the kidney medulla, consisting of a descending limb and an ascending limb. It is situated between the proximal kidney tubule and the distal kidney tubule.Tubular System, where 30% of filtered salt is normally reabsorbed. Bartter syndrome is characterized by salt wasting and hypokalemiaHypokalemiaHypokalemia is defined as plasma potassium (K+) concentration < 3.5 mEq/L. Homeostatic mechanisms maintain plasma concentration between 3.5-5.2 mEq/L despite marked variation in dietary intake. Hypokalemia can be due to renal losses, GI losses, transcellular shifts, or poor dietary intake.Hypokalemia and presents with electrolyte abnormalities and their consequences, such as vomitingVomitingThe forcible expulsion of the contents of the stomach through the mouth.Hypokalemia and dehydrationDehydrationThe condition that results from excessive loss of water from a living organism.Volume Depletion and Dehydration. Diagnosis is made by lab testing that shows the typical hypokalemic metabolic alkalosisAlkalosisA pathological condition that removes acid or adds base to the body fluids.Respiratory Alkalosis and hypercalciuriaHypercalciuriaExcretion of abnormally high level of calcium in the urine, greater than 4 mg/kg/day.Nephrolithiasis. Additional lab abnormalities include increased serum reninReninA highly specific (leu-leu) endopeptidase that generates angiotensin I from its precursor angiotensinogen, leading to a cascade of reactions which elevate blood pressure and increase sodium retention by the kidney in the renin-angiotensin system.Renal Sodium and Water Regulation and aldosteroneAldosteroneA hormone secreted by the adrenal cortex that regulates electrolyte and water balance by increasing the renal retention of sodium and the excretion of potassium.Hyperkalemia, but patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship clinically have normal blood pressure. Management is focused on normalizing serum electrolyte levels. ACE inhibitorsACE inhibitorsTruncus Arteriosus and angiotensin receptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors blockers are used to improve hypokalemiaHypokalemiaHypokalemia is defined as plasma potassium (K+) concentration < 3.5 mEq/L. Homeostatic mechanisms maintain plasma concentration between 3.5-5.2 mEq/L despite marked variation in dietary intake. Hypokalemia can be due to renal losses, GI losses, transcellular shifts, or poor dietary intake.Hypokalemia and limitLimitA value (e.g., pressure or time) that should not be exceeded and which is specified by the operator to protect the lungInvasive Mechanical VentilationproteinuriaProteinuriaThe presence of proteins in the urine, an indicator of kidney diseases.Nephrotic Syndrome in Children.
Bartter syndrome(BS) is a rare genetic (autosomal recessiveAutosomal recessiveAutosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal recessive diseases are only expressed when 2 copies of the recessive allele are inherited.Autosomal Recessive and Autosomal Dominant Inheritance) disorder that results from a defect in sodiumSodiumA member of the alkali group of metals. It has the atomic symbol na, atomic number 11, and atomic weight 23.Hyponatremiachloride reabsorptionChloride reabsorptionTubular System in the thick ascending limbThick ascending limbRenal Sodium and Water Regulation of the loop of HenleLoop of HenleThe U-shaped portion of the renal tubule in the kidney medulla, consisting of a descending limb and an ascending limb. It is situated between the proximal kidney tubule and the distal kidney tubule.Tubular System, leading to hypokalemiaHypokalemiaHypokalemia is defined as plasma potassium (K+) concentration < 3.5 mEq/L. Homeostatic mechanisms maintain plasma concentration between 3.5-5.2 mEq/L despite marked variation in dietary intake. Hypokalemia can be due to renal losses, GI losses, transcellular shifts, or poor dietary intake.Hypokalemia and metabolic alkalosisAlkalosisA pathological condition that removes acid or adds base to the body fluids.Respiratory Alkalosis. The disorder mimics long-term ingestion of a loop diuretic.
Epidemiology
PrevalencePrevalenceThe total number of cases of a given disease in a specified population at a designated time. It is differentiated from incidence, which refers to the number of new cases in the population at a given time.Measures of Disease Frequency: 1 in 1 million people in the United States
BS is less common than Gitelman syndromeGitelman syndromeGitelman syndrome is a rare genetic autosomal recessive disorder that affects the sodium-chloride cotransporter in the distal convoluted tubule of the nephron and causes electrolyte abnormalities. The syndrome presents clinically with symptoms of hypokalemia and hypomagnesemia. Gitelman Syndrome (a similar disorder of the renal tubule, which is seen in 1–10 in 40,000 people).
The prevalencePrevalenceThe total number of cases of a given disease in a specified population at a designated time. It is differentiated from incidence, which refers to the number of new cases in the population at a given time.Measures of Disease Frequency of heterozygotes with one of the genetic mutationsGenetic MutationsCarcinogenesis that cause BS is > 1% in the United States and as high as 3% in AsiaASIASpinal Cord Injuries.
Pathophysiology and Classification
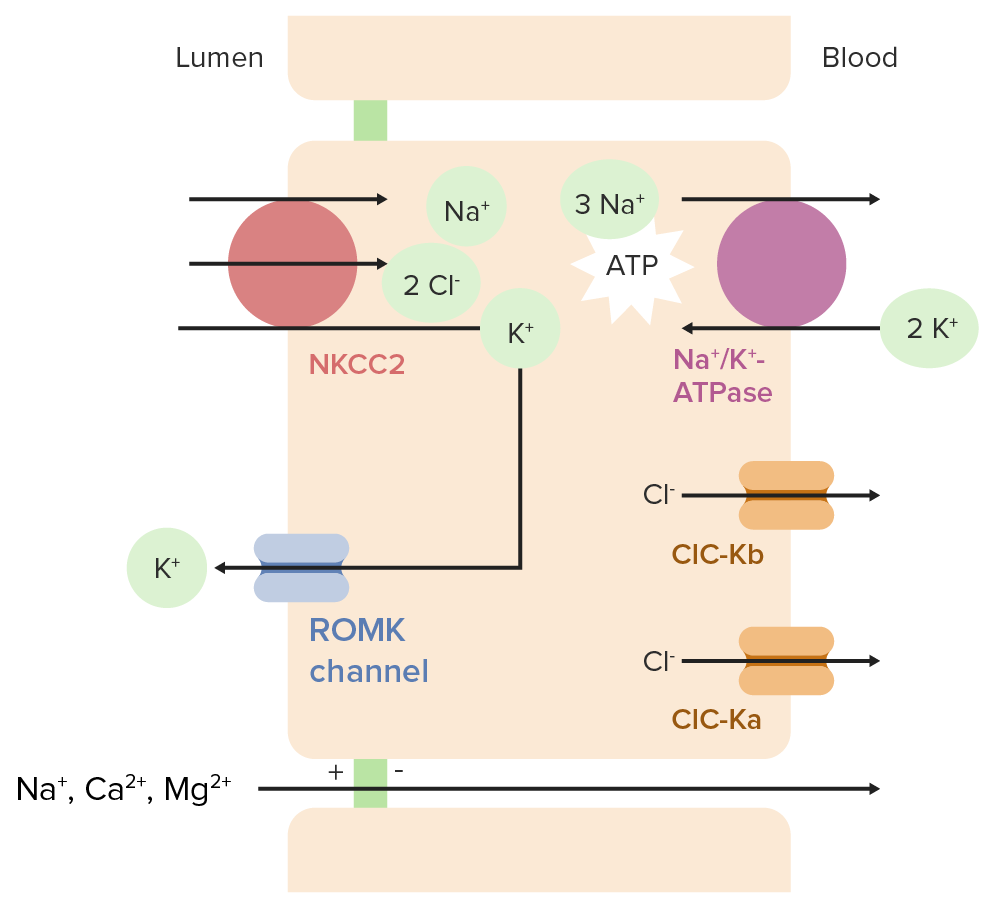
Normal physiology in the Loop of HenleLoop of HenleThe U-shaped portion of the renal tubule in the kidney medulla, consisting of a descending limb and an ascending limb. It is situated between the proximal kidney tubule and the distal kidney tubule.Tubular System
In the cells lining the loop of HenleLoop of HenleThe U-shaped portion of the renal tubule in the kidney medulla, consisting of a descending limb and an ascending limb. It is situated between the proximal kidney tubule and the distal kidney tubule.Tubular System:
The sodiumSodiumA member of the alkali group of metals. It has the atomic symbol na, atomic number 11, and atomic weight 23.Hyponatremia/potassiumPotassiumAn element in the alkali group of metals with an atomic symbol k, atomic number 19, and atomic weight 39. 10. It is the chief cation in the intracellular fluid of muscle and other cells. Potassium ion is a strong electrolyte that plays a significant role in the regulation of fluid volume and maintenance of the water-electrolyte balance.Hyperkalemia–adenosineAdenosineA nucleoside that is composed of adenine and d-ribose. Adenosine or adenosine derivatives play many important biological roles in addition to being components of DNA and RNA. Adenosine itself is a neurotransmitter.Class 5 Antiarrhythmic Drugs triphosphatase (Na+/K+-ATPase) pumpPumpACES and RUSH: Resuscitation Ultrasound Protocols on the basolateral membrane brings 3 Na+ out of the cell (into the blood) and 2 K+ into the cell, resulting in:
↓ Intracellular Na+ concentration
↑ Intracellular K+ concentration
The Na+/K+-2Cl cotransporter (also known as NKCC2NKCC2Renal Potassium Regulation) on the apical membrane reabsorbs 1 Na+, 1 K+, and 2 Cl– from the tubular lumen.
Electroneutral (2 positive and 2 negative charges moving in the same direction)
Driven by the low Na+ concentration within the cell
ClC-Ka (chlorideChlorideInorganic compounds derived from hydrochloric acid that contain the Cl- ion.Electrolytes channel Ka) is another chlorideChlorideInorganic compounds derived from hydrochloric acid that contain the Cl- ion.Electrolytes channel thought to have redundant functionality
A small protein subunit called barttin is required for both ClC-Kb and ClC-Ka to function properly
K+ that is brought into the cell through NKCC2NKCC2Renal Potassium Regulation is recycled back into the lumen through the renal outer medullary potassiumPotassiumAn element in the alkali group of metals with an atomic symbol k, atomic number 19, and atomic weight 39. 10. It is the chief cation in the intracellular fluid of muscle and other cells. Potassium ion is a strong electrolyte that plays a significant role in the regulation of fluid volume and maintenance of the water-electrolyte balance.Hyperkalemia (ROMK) channelsChannelsThe Cell: Cell Membrane → allows for continued NaCl reabsorption
CalciumCalciumA basic element found in nearly all tissues. It is a member of the alkaline earth family of metals with the atomic symbol ca, atomic number 20, and atomic weight 40. Calcium is the most abundant mineral in the body and combines with phosphorus to form calcium phosphate in the bones and teeth. It is essential for the normal functioning of nerves and muscles and plays a role in blood coagulation (as factor IV) and in many enzymatic processes.Electrolytes and magnesium reabsorptionMagnesium reabsorptionTubular System:
The movement of positively charged K+ into the lumen and negatively charged Cl– out of the lumen (and into the blood) causes the lumen to become more positively charged than the peritubular space.
This positive charge drives the paracellularParacellularRenal Potassium Regulation reabsorption of Na+, CaCACondylomata acuminata are a clinical manifestation of genital HPV infection. Condylomata acuminata are described as raised, pearly, flesh-colored, papular, cauliflower-like lesions seen in the anogenital region that may cause itching, pain, or bleeding.Condylomata Acuminata (Genital Warts)2+, and Mg2+.
Ion transport in the thick ascending limb of the loop of Henle ATPase: adenosine triphosphatase ClC-Ka: chloride channel Ka ClC-Kb: chloride channel Kb NKCC2: Na+/K+-Cl cotransporter 2 ROMK: renal outer medullary potassium
Image by Lecturio.
Pathophysiology in Bartter syndrome
Bartter syndrome can manifest from one of several autosomal recessiveAutosomal recessiveAutosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal recessive diseases are only expressed when 2 copies of the recessive allele are inherited.Autosomal Recessive and Autosomal Dominant Inheritance genetic defects. All subtypes are renal tubular disorders characterized by variableVariableVariables represent information about something that can change. The design of the measurement scales, or of the methods for obtaining information, will determine the data gathered and the characteristics of that data. As a result, a variable can be qualitative or quantitative, and may be further classified into subgroups.Types of Variables salt-wasting and hypokalemiaHypokalemiaHypokalemia is defined as plasma potassium (K+) concentration < 3.5 mEq/L. Homeostatic mechanisms maintain plasma concentration between 3.5-5.2 mEq/L despite marked variation in dietary intake. Hypokalemia can be due to renal losses, GI losses, transcellular shifts, or poor dietary intake.Hypokalemia.
SodiumSodiumA member of the alkali group of metals. It has the atomic symbol na, atomic number 11, and atomic weight 23.Hyponatremia
PotassiumPotassiumAn element in the alkali group of metals with an atomic symbol k, atomic number 19, and atomic weight 39. 10. It is the chief cation in the intracellular fluid of muscle and other cells. Potassium ion is a strong electrolyte that plays a significant role in the regulation of fluid volume and maintenance of the water-electrolyte balance.Hyperkalemia
ChlorideChlorideInorganic compounds derived from hydrochloric acid that contain the Cl- ion.Electrolytes
↓ Reabsorption of ions → leads to ↑ distal delivery of these ions
These ions remain in the tubular lumen as the urine travels distally
↑ Distal delivery of Na+ → ↑ the electronegative gradient across the luminal membrane → ↑ excretion of K+ → hypokalemiaHypokalemiaHypokalemia is defined as plasma potassium (K+) concentration < 3.5 mEq/L. Homeostatic mechanisms maintain plasma concentration between 3.5-5.2 mEq/L despite marked variation in dietary intake. Hypokalemia can be due to renal losses, GI losses, transcellular shifts, or poor dietary intake.Hypokalemia
↑ Distal delivery of K+ → ↑ exchange of K+ for H+ in the collected duct → ↑ H+ excretion → metabolic alkalosisAlkalosisA pathological condition that removes acid or adds base to the body fluids.Respiratory Alkalosis
↑ Na+ in the urine → ↑ free water in the urine, resulting in:
Impaired ability to concentrate the urine
Volume depletionVolume depletionVolume status is a balance between water and solutes, the majority of which is Na. Volume depletion refers to a loss of both water and Na, whereas dehydration refers only to a loss of water. Volume depletion can be caused by GI losses, renal losses, bleeding, poor oral Na intake, or third spacing of fluids.Volume Depletion and Dehydration → causes activation of the RAASRAASA blood pressure regulating system of interacting components that include renin; angiotensinogen; angiotensin converting enzyme; angiotensin i; angiotensin ii; and angiotensinase. Renin, an enzyme produced in the kidney, acts on angiotensinogen, an alpha-2 globulin produced by the liver, forming angiotensin I. Angiotensin-converting enzyme, contained in the lung, acts on angiotensin I in the plasma converting it to angiotensin II, an extremely powerful vasoconstrictor. Angiotensin II causes contraction of the arteriolar and renal vascular smooth muscle, leading to retention of salt and water in the kidney and increased arterial blood pressure. In addition, angiotensin II stimulates the release of aldosterone from the adrenal cortex, which in turn also increases salt and water retention in the kidney. Angiotensin-converting enzyme also breaks down bradykinin, a powerful vasodilator and component of the kallikrein-kinin system.Adrenal Hormones and leads to secondary hyperaldosteronismSecondary hyperaldosteronismPhysiological oversecretion of aldosterone that occurs in response to overstimulation of the RAAS, triggered by decreases in renal blood flow.Hyperaldosteronism
Long-term stimulation of the RAASRAASA blood pressure regulating system of interacting components that include renin; angiotensinogen; angiotensin converting enzyme; angiotensin i; angiotensin ii; and angiotensinase. Renin, an enzyme produced in the kidney, acts on angiotensinogen, an alpha-2 globulin produced by the liver, forming angiotensin I. Angiotensin-converting enzyme, contained in the lung, acts on angiotensin I in the plasma converting it to angiotensin II, an extremely powerful vasoconstrictor. Angiotensin II causes contraction of the arteriolar and renal vascular smooth muscle, leading to retention of salt and water in the kidney and increased arterial blood pressure. In addition, angiotensin II stimulates the release of aldosterone from the adrenal cortex, which in turn also increases salt and water retention in the kidney. Angiotensin-converting enzyme also breaks down bradykinin, a powerful vasodilator and component of the kallikrein-kinin system.Adrenal Hormones causes hyperplasiaHyperplasiaAn increase in the number of cells in a tissue or organ without tumor formation. It differs from hypertrophy, which is an increase in bulk without an increase in the number of cells.Cellular Adaptation of the juxtaglomerular apparatus and increased reninReninA highly specific (leu-leu) endopeptidase that generates angiotensin I from its precursor angiotensinogen, leading to a cascade of reactions which elevate blood pressure and increase sodium retention by the kidney in the renin-angiotensin system.Renal Sodium and Water Regulation levels.
↑ Renal release of prostaglandin E2Prostaglandin E2The most common and most biologically active of the mammalian prostaglandins. It exhibits most biological activities characteristic of prostaglandins and has been used extensively as an oxytocic agent. The compound also displays a protective effect on the intestinal mucosa.Fever
↑ Renal blood flowRenal blood flowThe amount of the renal blood flow that is going to the functional renal tissue, i.e., parts of the kidney that are involved in production of urine.Glomerular Filtration and GFRGFRThe volume of water filtered out of plasma through glomerular capillary walls into Bowman’s capsules per unit of time. It is considered to be equivalent to inulin clearance.Kidney Function Tests
↑ ReninReninA highly specific (leu-leu) endopeptidase that generates angiotensin I from its precursor angiotensinogen, leading to a cascade of reactions which elevate blood pressure and increase sodium retention by the kidney in the renin-angiotensin system.Renal Sodium and Water RegulationsecretionSecretionCoagulation Studies
↑ Na+ and free water excretion
Also ↑ urinary loss of calciumCalciumA basic element found in nearly all tissues. It is a member of the alkaline earth family of metals with the atomic symbol ca, atomic number 20, and atomic weight 40. Calcium is the most abundant mineral in the body and combines with phosphorus to form calcium phosphate in the bones and teeth. It is essential for the normal functioning of nerves and muscles and plays a role in blood coagulation (as factor IV) and in many enzymatic processes.Electrolytes and magnesiumMagnesiumA metallic element that has the atomic symbol mg, atomic number 12, and atomic weight 24. 31. It is important for the activity of many enzymes, especially those involved in oxidative phosphorylation.Electrolytes.
↓ NaCl reabsorption → ↓ CaCACondylomata acuminata are a clinical manifestation of genital HPV infection. Condylomata acuminata are described as raised, pearly, flesh-colored, papular, cauliflower-like lesions seen in the anogenital region that may cause itching, pain, or bleeding.Condylomata Acuminata (Genital Warts)2+ and Mg+ reabsorption
CalciumCalciumA basic element found in nearly all tissues. It is a member of the alkaline earth family of metals with the atomic symbol ca, atomic number 20, and atomic weight 40. Calcium is the most abundant mineral in the body and combines with phosphorus to form calcium phosphate in the bones and teeth. It is essential for the normal functioning of nerves and muscles and plays a role in blood coagulation (as factor IV) and in many enzymatic processes.Electrolytes wasting in the urine can lead to nephrocalcinosisNephrocalcinosisA condition characterized by calcification of the renal tissue itself. It is usually seen in distal renal tubular acidosis with calcium deposition in the distal kidney tubules and the surrounding interstitium. Nephrocalcinosis causes renal insufficiency.X-linked Hypophosphatemic Rickets.
Activating mutations in the calcium-sensing receptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors (CaSR) on the basolateral membrane can also impair NaCl transport → generates a mild form of BS
This mutationMutationGenetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations is autosomal dominantAutosomal dominantAutosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal dominant diseases are expressed when only 1 copy of the dominant allele is inherited. Autosomal Recessive and Autosomal Dominant Inheritance.
Also leads to a downward “resetting” of the normal serum calciumCalciumA basic element found in nearly all tissues. It is a member of the alkaline earth family of metals with the atomic symbol ca, atomic number 20, and atomic weight 40. Calcium is the most abundant mineral in the body and combines with phosphorus to form calcium phosphate in the bones and teeth. It is essential for the normal functioning of nerves and muscles and plays a role in blood coagulation (as factor IV) and in many enzymatic processes.Electrolytes range → results in ↓ parathyroidParathyroidThe parathyroid glands are 2 pairs of small endocrine glands found in close proximity to the thyroid gland. The superior parathyroid glands are lodged within the parenchyma of the upper poles of the right and left thyroid lobes; the inferior parathyroid glands are close to the inferior tips or poles of the lobes.Parathyroid Glands: Anatomy hormone (PTH) and hypocalcemiaHypocalcemiaHypocalcemia, a serum calcium < 8.5 mg/dL, can result from various conditions. The causes may include hypoparathyroidism, drugs, disorders leading to vitamin D deficiency, and more. Calcium levels are regulated and affected by different elements such as dietary intake, parathyroid hormone (PTH), vitamin D, pH, and albumin. Presentation can range from an asymptomatic (mild deficiency) to a life-threatening condition (acute, significant deficiency). Hypocalcemia
Classification by mutations
There is significant genetic heterogeneity in BS; it may result from homozygous or mixed heterozygous mutations in any of the genesGenesA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.DNA Types and Structure that reduce the activity of electrolyte transporters in the TALTALRenal Sodium and Water Regulation. Thus, the severity and clinical presentation of BS vary with each type.
Note that Autosomal dominantAutosomal dominantAutosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal dominant diseases are expressed when only 1 copy of the dominant allele is inherited. Autosomal Recessive and Autosomal Dominant InheritancehypocalcemiaHypocalcemiaHypocalcemia, a serum calcium < 8.5 mg/dL, can result from various conditions. The causes may include hypoparathyroidism, drugs, disorders leading to vitamin D deficiency, and more. Calcium levels are regulated and affected by different elements such as dietary intake, parathyroid hormone (PTH), vitamin D, pH, and albumin. Presentation can range from an asymptomatic (mild deficiency) to a life-threatening condition (acute, significant deficiency). Hypocalcemia, a condition associated with an activating mutationMutationGenetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations of the CASRgeneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics (affecting the calcium-sensing receptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors), was previously classified as Type V Bartter syndrome. Some cases can have potassiumPotassiumAn element in the alkali group of metals with an atomic symbol k, atomic number 19, and atomic weight 39. 10. It is the chief cation in the intracellular fluid of muscle and other cells. Potassium ion is a strong electrolyte that plays a significant role in the regulation of fluid volume and maintenance of the water-electrolyte balance.Hyperkalemia wasting and metabolic alkalosisAlkalosisA pathological condition that removes acid or adds base to the body fluids.Respiratory Alkalosis, which is similar to Bartter syndrome.
Type V BS is transient antenatal Bartter syndrome, a severe but transient form. It is associated with mutations in MAGED2, a geneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics on the X chromosomeX chromosomeThe female sex chromosome, being the differential sex chromosome carried by half the male gametes and all female gametes in human and other male-heterogametic species.Basic Terms of Genetics encoding melanoma-associated antigenAntigenSubstances that are recognized by the immune system and induce an immune reaction.Vaccination D2 (MAGE-D2). This affects fetal renal salt reabsorption and presentation includes polyhydramniosPolyhydramniosPolyhydramnios is a pathological excess of amniotic fluid. Common causes of polyhydramnios include fetal anomalies, gestational diabetes, multiple gestations, and congenital infections. Patients are often asymptomatic but may present with dyspnea, extremity swelling, or abdominal distention. Polyhydramnios, prematurityPrematurityNeonatal Respiratory Distress Syndrome, hyponatremiaHyponatremiaHyponatremia is defined as a decreased serum sodium (sNa+) concentration less than 135 mmol/L. Serum sodium is the greatest contributor to plasma osmolality, which is very tightly controlled via antidiuretic hormone (ADH) release from the hypothalamus and by the thirst mechanism.Hyponatremia, metabolic alkalosisAlkalosisA pathological condition that removes acid or adds base to the body fluids.Respiratory Alkalosis and polyuriaPolyuriaUrination of a large volume of urine with an increase in urinary frequency, commonly seen in diabetes.Renal Potassium Regulation.
Table: Summary of 5 Bartter syndrome subtypes depending on the geneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics involved and the transporter being affected
Name
Type
Defective protein
Severity of presentation
Neonatal Bartter syndrome (hyperprostaglandin E syndrome)
Classic Bartter syndrome with sensorineural deafness (hyperprostaglandin E syndrome)
IVa
Barttin (the β-subunit of ClC-Ka and ClC-Kb)
Severe
IVb
Simultaneous mutations in ClC-Ka and ClC-Kb
Severe
Bartter syndrome with hypocalcemiaHypocalcemiaHypocalcemia, a serum calcium < 8.5 mg/dL, can result from various conditions. The causes may include hypoparathyroidism, drugs, disorders leading to vitamin D deficiency, and more. Calcium levels are regulated and affected by different elements such as dietary intake, parathyroid hormone (PTH), vitamin D, pH, and albumin. Presentation can range from an asymptomatic (mild deficiency) to a life-threatening condition (acute, significant deficiency). Hypocalcemia (also called autosomal dominantAutosomal dominantAutosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal dominant diseases are expressed when only 1 copy of the dominant allele is inherited. Autosomal Recessive and Autosomal Dominant InheritancehypoparathyroidismHypoparathyroidismHypoparathyroidism is defined as reduced parathyroid hormone (PTH) levels due to poor function of the parathyroid glands. The cause of hypoparathyroidism is most commonly iatrogenic following neck surgery, but it can also be associated with genetic or autoimmune disorders as well as infiltrative diseases causing destruction of the normal parathyroid tissue. Hypoparathyroidism)
V
CaSR
Mild
Different channelopathies in Bartter’s syndrome ATPase: adenosine triphosphatase ClC-Ka: chloride channel Ka ClC-Kb: chloride channel Kb NKCC2: Na+/K+-Cl cotransporter 2 ROMK: renal outer medullary potassium
The clinical manifestations of BS are mostly due to electrolyte imbalances and their consequences. Symptoms are much less pronounced in heterozygotes. The tubular defects in ion transport produce a clinical disorder that appears similar to that seen with long-term ingestion of a loop diuretic (e.g., furosemideFurosemideA benzoic-sulfonamide-furan. It is a diuretic with fast onset and short duration that is used for edema and chronic renal insufficiency.Loop Diuretics).
Presentations in neonates
Typically seen in types I, II, IVa, and IVb. Common findings include:
PolyhydramniosPolyhydramniosPolyhydramnios is a pathological excess of amniotic fluid. Common causes of polyhydramnios include fetal anomalies, gestational diabetes, multiple gestations, and congenital infections. Patients are often asymptomatic but may present with dyspnea, extremity swelling, or abdominal distention. Polyhydramnios antenatally
Preterm birthPreterm birthPreterm labor refers to regular uterine contractions leading to cervical change prior to 37 weeks of gestation; preterm birth refers to birth prior to 37 weeks of gestation. Preterm birth may be spontaneous due to preterm labor, preterm prelabor rupture of membranes (PPROM), or cervical insufficiency. Preterm Labor and Birth
Sensorineural deafness (types IVa and IVb)
Electrolyte abnormalities:
HypokalemiaHypokalemiaHypokalemia is defined as plasma potassium (K+) concentration < 3.5 mEq/L. Homeostatic mechanisms maintain plasma concentration between 3.5-5.2 mEq/L despite marked variation in dietary intake. Hypokalemia can be due to renal losses, GI losses, transcellular shifts, or poor dietary intake.Hypokalemia
Metabolic alkalosisAlkalosisA pathological condition that removes acid or adds base to the body fluids.Respiratory Alkalosis
HypercalciuriaHypercalciuriaExcretion of abnormally high level of calcium in the urine, greater than 4 mg/kg/day.Nephrolithiasis
PolyuriaPolyuriaUrination of a large volume of urine with an increase in urinary frequency, commonly seen in diabetes.Renal Potassium Regulation
VomitingVomitingThe forcible expulsion of the contents of the stomach through the mouth.Hypokalemia
DiarrheaDiarrheaDiarrhea is defined as ≥ 3 watery or loose stools in a 24-hour period. There are a multitude of etiologies, which can be classified based on the underlying mechanism of disease. The duration of symptoms (acute or chronic) and characteristics of the stools (e.g., watery, bloody, steatorrheic, mucoid) can help guide further diagnostic evaluation. Diarrhea
EmaciationEmaciationClinical manifestation of excessive leanness usually caused by disease or a lack of nutrition (malnutrition).Anorexia Nervosa/failure to thriveFailure to ThriveFailure to thrive (FTT), or faltering growth, describes suboptimal weight gain and growth in children. The majority of cases are due to inadequate caloric intake; however, genetic, infectious, and oncological etiologies are also common. Failure to Thrive
Growth and developmental delay
Abnormal facial features
Prominent foreheadForeheadThe part of the face above the eyes.Melasma
Large eyes
StrabismusStrabismusStrabismus is the misalignment of the eyes while fixating the gaze on an object. Strabismus can be idiopathic, but it may also be caused by cerebral palsy, uncorrected refractive errors, and extraocular muscle or cranial nerve dysfunction. Strabismus
Protruding ears
Drooping mouth
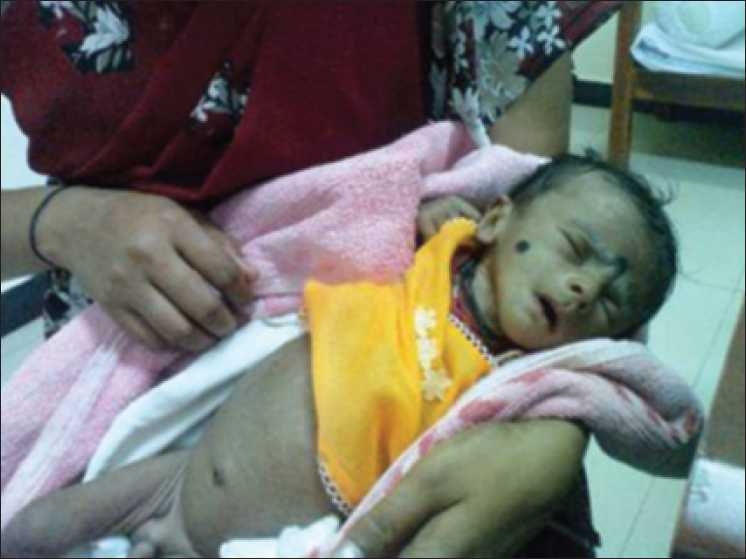
Child with Bartter syndrome showing severe dehydration before treatment
Image: “Bartter’s child before treatment showing severe dehydration” by Sampathkumar K et al. License: CC BY 2.0
Presentations in children, adolescents, and adults
Common findings include:
Electrolyte abnormalities:
HypokalemiaHypokalemiaHypokalemia is defined as plasma potassium (K+) concentration < 3.5 mEq/L. Homeostatic mechanisms maintain plasma concentration between 3.5-5.2 mEq/L despite marked variation in dietary intake. Hypokalemia can be due to renal losses, GI losses, transcellular shifts, or poor dietary intake.Hypokalemia
Metabolic alkalosisAlkalosisA pathological condition that removes acid or adds base to the body fluids.Respiratory Alkalosis
HypercalciuriaHypercalciuriaExcretion of abnormally high level of calcium in the urine, greater than 4 mg/kg/day.Nephrolithiasis
Hypophosphatemia in occasional patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship with secondary hyperparathyroidismSecondary hyperparathyroidismAbnormally elevated parathyroid hormone secretion as a response to hypocalcemia. It is caused by chronic kidney failure or other abnormalities in the controls of bone and mineral metabolism, leading to various bone diseases, such as renal osteodystrophy.Hyperparathyroidism
Normal or mildly decreased serum magnesiumMagnesiumA metallic element that has the atomic symbol mg, atomic number 12, and atomic weight 24. 31. It is important for the activity of many enzymes, especially those involved in oxidative phosphorylation.Electrolytes level
VomitingVomitingThe forcible expulsion of the contents of the stomach through the mouth.Hypokalemia
PolyuriaPolyuriaUrination of a large volume of urine with an increase in urinary frequency, commonly seen in diabetes.Renal Potassium Regulation and polydipsiaPolydipsiaExcessive thirst manifested by excessive fluid intake. It is characteristic of many diseases such as diabetes mellitus; diabetes insipidus; and nephrogenic diabetes insipidus. The condition may be psychogenic in origin.Arginine Vasopressin Disorders (Diabetes Insipidus) due to decreased urinary concentrating ability
HypotensionHypotensionHypotension is defined as low blood pressure, specifically < 90/60 mm Hg, and is most commonly a physiologic response. Hypotension may be mild, serious, or life threatening, depending on the cause. Hypotension
ConstipationConstipationConstipation is common and may be due to a variety of causes. Constipation is generally defined as bowel movement frequency < 3 times per week. Patients who are constipated often strain to pass hard stools. The condition is classified as primary (also known as idiopathic or functional constipation) or secondary, and as acute or chronic. Constipation
↓ GFRGFRThe volume of water filtered out of plasma through glomerular capillary walls into Bowman’s capsules per unit of time. It is considered to be equivalent to inulin clearance.Kidney Function Tests
Diagnosis
The diagnosis of BS is made by lab findings after clinical suspicion arises from the history and physical examination.
History and exam
Evaluate for surreptitious vomitingVomitingThe forcible expulsion of the contents of the stomach through the mouth.Hypokalemia; findings may include:
ScarringScarringInflammation on the handHandThe hand constitutes the distal part of the upper limb and provides the fine, precise movements needed in activities of daily living. It consists of 5 metacarpal bones and 14 phalanges, as well as numerous muscles innervated by the median and ulnar nerves. Hand: Anatomy from insertion into the mouth
Ask about family historyFamily HistoryAdult Health Maintenance of nephrocalcinosisNephrocalcinosisA condition characterized by calcification of the renal tissue itself. It is usually seen in distal renal tubular acidosis with calcium deposition in the distal kidney tubules and the surrounding interstitium. Nephrocalcinosis causes renal insufficiency.X-linked Hypophosphatemic Rickets.
Look for characteristic facial features.
HypotensionHypotensionHypotension is defined as low blood pressure, specifically < 90/60 mm Hg, and is most commonly a physiologic response. Hypotension may be mild, serious, or life threatening, depending on the cause. Hypotension (Primary hyperaldosteronismPrimary hyperaldosteronismAutonomous (renin-independent) secretion of aldosterone.Hyperaldosteronism will typically present with hypertensionHypertensionHypertension, or high blood pressure, is a common disease that manifests as elevated systemic arterial pressures. Hypertension is most often asymptomatic and is found incidentally as part of a routine physical examination or during triage for an unrelated medical encounter. Hypertension.)
Laboratory
Serum electrolytesElectrolytesElectrolytes are mineral salts that dissolve in water and dissociate into charged particles called ions, which can be either be positively (cations) or negatively (anions) charged. Electrolytes are distributed in the extracellular and intracellular compartments in different concentrations. Electrolytes are essential for various basic life-sustaining functions.Electrolytes:
Unexplained hypokalemiaHypokalemiaHypokalemia is defined as plasma potassium (K+) concentration < 3.5 mEq/L. Homeostatic mechanisms maintain plasma concentration between 3.5-5.2 mEq/L despite marked variation in dietary intake. Hypokalemia can be due to renal losses, GI losses, transcellular shifts, or poor dietary intake.Hypokalemia (a key diagnostic feature of BS)
Metabolic alkalosisAlkalosisA pathological condition that removes acid or adds base to the body fluids.Respiratory Alkalosis
HypomagnesemiaHypomagnesemiaA nutritional condition produced by a deficiency of magnesium in the diet, characterized by anorexia, nausea, vomiting, lethargy, and weakness. Symptoms are paresthesias, muscle cramps, irritability, decreased attention span, and mental confusion, possibly requiring months to appear. Deficiency of body magnesium can exist even when serum values are normal. In addition, magnesium deficiency may be organ-selective, since certain tissues become deficient before others. Electrolytes
Hypophosphatemia
Urine electrolytesElectrolytesElectrolytes are mineral salts that dissolve in water and dissociate into charged particles called ions, which can be either be positively (cations) or negatively (anions) charged. Electrolytes are distributed in the extracellular and intracellular compartments in different concentrations. Electrolytes are essential for various basic life-sustaining functions.Electrolytes:
↑ Urinary chlorideChlorideInorganic compounds derived from hydrochloric acid that contain the Cl- ion.Electrolytes (differentiates BS from surreptitious vomitingVomitingThe forcible expulsion of the contents of the stomach through the mouth.Hypokalemia, which will have a ↓ urine Cl–)
↑ Urinary calciumCalciumA basic element found in nearly all tissues. It is a member of the alkaline earth family of metals with the atomic symbol ca, atomic number 20, and atomic weight 40. Calcium is the most abundant mineral in the body and combines with phosphorus to form calcium phosphate in the bones and teeth. It is essential for the normal functioning of nerves and muscles and plays a role in blood coagulation (as factor IV) and in many enzymatic processes.Electrolytes (differentiates BS from Gitelman syndromeGitelman syndromeGitelman syndrome is a rare genetic autosomal recessive disorder that affects the sodium-chloride cotransporter in the distal convoluted tubule of the nephron and causes electrolyte abnormalities. The syndrome presents clinically with symptoms of hypokalemia and hypomagnesemia. Gitelman Syndrome, which will have a ↓ urine CaCACondylomata acuminata are a clinical manifestation of genital HPV infection. Condylomata acuminata are described as raised, pearly, flesh-colored, papular, cauliflower-like lesions seen in the anogenital region that may cause itching, pain, or bleeding.Condylomata Acuminata (Genital Warts)2+)
↑ Urinary sodiumSodiumA member of the alkali group of metals. It has the atomic symbol na, atomic number 11, and atomic weight 23.Hyponatremia
↑ Urinary potassiumPotassiumAn element in the alkali group of metals with an atomic symbol k, atomic number 19, and atomic weight 39. 10. It is the chief cation in the intracellular fluid of muscle and other cells. Potassium ion is a strong electrolyte that plays a significant role in the regulation of fluid volume and maintenance of the water-electrolyte balance.Hyperkalemia
Additional lab abnormalities:
↑ Serum reninReninA highly specific (leu-leu) endopeptidase that generates angiotensin I from its precursor angiotensinogen, leading to a cascade of reactions which elevate blood pressure and increase sodium retention by the kidney in the renin-angiotensin system.Renal Sodium and Water Regulation
↑ Serum aldosteroneAldosteroneA hormone secreted by the adrenal cortex that regulates electrolyte and water balance by increasing the renal retention of sodium and the excretion of potassium.Hyperkalemia
↑ Prostaglandin E2Prostaglandin E2The most common and most biologically active of the mammalian prostaglandins. It exhibits most biological activities characteristic of prostaglandins and has been used extensively as an oxytocic agent. The compound also displays a protective effect on the intestinal mucosa.Fever
↑ PTH
Genetic testingGenetic TestingDetection of a mutation; genotype; karyotype; or specific alleles associated with genetic traits, heritable diseases, or predisposition to a disease, or that may lead to the disease in descendants. It includes prenatal genetic testing.Myotonic Dystrophies: gold standard; identifies specific mutations
Prenatal testing (not routinely used):
Amniotic fluidAmniotic fluidA clear, yellowish liquid that envelopes the fetus inside the sac of amnion. In the first trimester, it is likely a transudate of maternal or fetal plasma. In the second trimester, amniotic fluid derives primarily from fetal lung and kidney. Cells or substances in this fluid can be removed for prenatal diagnostic tests (amniocentesis).Placenta, Umbilical Cord, and Amniotic CavitychlorideChlorideInorganic compounds derived from hydrochloric acid that contain the Cl- ion.Electrolytes levels may be elevated.
The tubular defects in BS cannotbe corrected (except by kidney transplantationKidney TransplantationThe transference of a kidney from one human or animal to another.Organ Transplantation). The goal of management is to decrease the effects of elevated prostaglandinsProstaglandinsA group of compounds derived from unsaturated 20-carbon fatty acids, primarily arachidonic acid, via the cyclooxygenase pathway. They are extremely potent mediators of a diverse group of physiological processes.Eicosanoids, reninReninA highly specific (leu-leu) endopeptidase that generates angiotensin I from its precursor angiotensinogen, leading to a cascade of reactions which elevate blood pressure and increase sodium retention by the kidney in the renin-angiotensin system.Renal Sodium and Water Regulation, and angiotensin in types I, II, and IV. In the milder adult form, or classic BS, the primary goal is to normalize serum potassiumPotassiumAn element in the alkali group of metals with an atomic symbol k, atomic number 19, and atomic weight 39. 10. It is the chief cation in the intracellular fluid of muscle and other cells. Potassium ion is a strong electrolyte that plays a significant role in the regulation of fluid volume and maintenance of the water-electrolyte balance.Hyperkalemia levels.
Prenatal Bartter syndrome with severe polyhydramniosPolyhydramniosPolyhydramnios is a pathological excess of amniotic fluid. Common causes of polyhydramnios include fetal anomalies, gestational diabetes, multiple gestations, and congenital infections. Patients are often asymptomatic but may present with dyspnea, extremity swelling, or abdominal distention. Polyhydramnios
NSAIDsNSAIDSPrimary vs Secondary Headaches to antagonize the effects of the ↑ prostaglandinsProstaglandinsA group of compounds derived from unsaturated 20-carbon fatty acids, primarily arachidonic acid, via the cyclooxygenase pathway. They are extremely potent mediators of a diverse group of physiological processes.Eicosanoids
Consider intermittent amniocentesisAmniocentesisPercutaneous transabdominal puncture of the uterus during pregnancy to obtain amniotic fluid. It is commonly used for fetal karyotype determination in order to diagnose abnormal fetal conditions.Polyhydramnios in the 3rd trimester to treat severe polyhydramniosPolyhydramniosPolyhydramnios is a pathological excess of amniotic fluid. Common causes of polyhydramnios include fetal anomalies, gestational diabetes, multiple gestations, and congenital infections. Patients are often asymptomatic but may present with dyspnea, extremity swelling, or abdominal distention. Polyhydramnios by draining excessive amniotic fluidAmniotic fluidA clear, yellowish liquid that envelopes the fetus inside the sac of amnion. In the first trimester, it is likely a transudate of maternal or fetal plasma. In the second trimester, amniotic fluid derives primarily from fetal lung and kidney. Cells or substances in this fluid can be removed for prenatal diagnostic tests (amniocentesis).Placenta, Umbilical Cord, and Amniotic Cavity.
Neonatal Bartter syndrome types I, II, and IV
IV saline infusion may be needed to treat dehydrationDehydrationThe condition that results from excessive loss of water from a living organism.Volume Depletion and Dehydration.
Oral potassiumPotassiumAn element in the alkali group of metals with an atomic symbol k, atomic number 19, and atomic weight 39. 10. It is the chief cation in the intracellular fluid of muscle and other cells. Potassium ion is a strong electrolyte that plays a significant role in the regulation of fluid volume and maintenance of the water-electrolyte balance.Hyperkalemia supplementation will likely needed.
Childhood or adult Bartter syndrome type III
Oral supplementation of electrolytesElectrolytesElectrolytes are mineral salts that dissolve in water and dissociate into charged particles called ions, which can be either be positively (cations) or negatively (anions) charged. Electrolytes are distributed in the extracellular and intracellular compartments in different concentrations. Electrolytes are essential for various basic life-sustaining functions.Electrolytes:
To correct fluid and electrolyte imbalances
Most likely to require:
PotassiumPotassiumAn element in the alkali group of metals with an atomic symbol k, atomic number 19, and atomic weight 39. 10. It is the chief cation in the intracellular fluid of muscle and other cells. Potassium ion is a strong electrolyte that plays a significant role in the regulation of fluid volume and maintenance of the water-electrolyte balance.Hyperkalemia
SodiumSodiumA member of the alkali group of metals. It has the atomic symbol na, atomic number 11, and atomic weight 23.Hyponatremia
MagnesiumMagnesiumA metallic element that has the atomic symbol mg, atomic number 12, and atomic weight 24. 31. It is important for the activity of many enzymes, especially those involved in oxidative phosphorylation.Electrolytes
Antagonize the effects of increased prostaglandinsProstaglandinsA group of compounds derived from unsaturated 20-carbon fatty acids, primarily arachidonic acid, via the cyclooxygenase pathway. They are extremely potent mediators of a diverse group of physiological processes.Eicosanoids
Examples: indomethacinIndomethacinA non-steroidal anti-inflammatory agent (nsaid) that inhibits cyclooxygenase, which is necessary for the formation of prostaglandins and other autacoids. It also inhibits the motility of polymorphonuclear leukocytes.Nonsteroidal Antiinflammatory Drugs (NSAIDs), celecoxibCelecoxibA pyrazole derivative and selective cyclooxygenase 2 inhibitor that is used to treat symptoms associated with rheumatoid arthritis; osteoarthritis and juvenile arthritis, as well as the management of acute pain.Nonsteroidal Antiinflammatory Drugs (NSAIDs)
Blocks distal tubule sodiumSodiumA member of the alkali group of metals. It has the atomic symbol na, atomic number 11, and atomic weight 23.Hyponatremia–potassiumPotassiumAn element in the alkali group of metals with an atomic symbol k, atomic number 19, and atomic weight 39. 10. It is the chief cation in the intracellular fluid of muscle and other cells. Potassium ion is a strong electrolyte that plays a significant role in the regulation of fluid volume and maintenance of the water-electrolyte balance.Hyperkalemia exchange → can ↑ serum K+ and reverse metabolic alkalosisAlkalosisA pathological condition that removes acid or adds base to the body fluids.Respiratory Alkalosis
Examples: amilorideAmilorideA pyrazine compound inhibiting sodium reabsorption through sodium channels in renal epithelial cells. This inhibition creates a negative potential in the luminal membranes of principal cells, located in the distal convoluted tubule and collecting duct. Negative potential reduces secretion of potassium and hydrogen ions. Amiloride is used in conjunction with diuretics to spare potassium loss.Liddle Syndrome, spironolactoneSpironolactoneA potassium sparing diuretic that acts by antagonism of aldosterone in the distal renal tubules. It is used mainly in the treatment of refractory edema in patients with congestive heart failure, nephrotic syndrome, or hepatic cirrhosis. Its effects on the endocrine system are utilized in the treatments of hirsutism and acne but they can lead to adverse effects.Potassium-sparing Diuretics
ACE inhibitorsACE inhibitorsTruncus Arteriosus and angiotensin receptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors blockers:
Decrease elevated angiotensin IIAngiotensin IIAn octapeptide that is a potent but labile vasoconstrictor. It is produced from angiotensin I after the removal of two amino acids at the c-terminal by angiotensin converting enzyme. The amino acid in position 5 varies in different species. To block vasoconstriction and hypertension effect of angiotensin II, patients are often treated with ace inhibitors or with angiotensin II type 1 receptor blockers.Renal Sodium and Water Regulation and aldosteroneAldosteroneA hormone secreted by the adrenal cortex that regulates electrolyte and water balance by increasing the renal retention of sodium and the excretion of potassium.Hyperkalemia levels
Increase serum potassiumPotassiumAn element in the alkali group of metals with an atomic symbol k, atomic number 19, and atomic weight 39. 10. It is the chief cation in the intracellular fluid of muscle and other cells. Potassium ion is a strong electrolyte that plays a significant role in the regulation of fluid volume and maintenance of the water-electrolyte balance.Hyperkalemia
For rare patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship with end-stage renal disease and/or nephrocalcinosisNephrocalcinosisA condition characterized by calcification of the renal tissue itself. It is usually seen in distal renal tubular acidosis with calcium deposition in the distal kidney tubules and the surrounding interstitium. Nephrocalcinosis causes renal insufficiency.X-linked Hypophosphatemic Rickets
Tubular abnormalities resolve after kidney transplantationKidney TransplantationThe transference of a kidney from one human or animal to another.Organ Transplantation, without recurrence.
Complications
Cardiac arrhythmias and sudden cardiac deathSudden cardiac deathCardiac arrest is the sudden, complete cessation of cardiac output with hemodynamic collapse. Patients present as pulseless, unresponsive, and apneic. Rhythms associated with cardiac arrest are ventricular fibrillation/tachycardia, asystole, or pulseless electrical activity.Cardiac Arrest due to electrolyte imbalances
Failure to thriveFailure to ThriveFailure to thrive (FTT), or faltering growth, describes suboptimal weight gain and growth in children. The majority of cases are due to inadequate caloric intake; however, genetic, infectious, and oncological etiologies are also common. Failure to Thrive
Developmental delay
OsteopeniaOsteopeniaOsteoporosis or osteoporosisOsteoporosisOsteoporosis refers to a decrease in bone mass and density leading to an increased number of fractures. There are 2 forms of osteoporosis: primary, which is commonly postmenopausal or senile; and secondary, which is a manifestation of immobilization, underlying medical disorders, or long-term use of certain medications. Osteoporosis due to calciumCalciumA basic element found in nearly all tissues. It is a member of the alkaline earth family of metals with the atomic symbol ca, atomic number 20, and atomic weight 40. Calcium is the most abundant mineral in the body and combines with phosphorus to form calcium phosphate in the bones and teeth. It is essential for the normal functioning of nerves and muscles and plays a role in blood coagulation (as factor IV) and in many enzymatic processes.Electrolytes loss in boneBoneBone is a compact type of hardened connective tissue composed of bone cells, membranes, an extracellular mineralized matrix, and central bone marrow. The 2 primary types of bone are compact and spongy. Bones: Structure and Types
Differential Diagnosis
Gitelman syndromeGitelman syndromeGitelman syndrome is a rare genetic autosomal recessive disorder that affects the sodium-chloride cotransporter in the distal convoluted tubule of the nephron and causes electrolyte abnormalities. The syndrome presents clinically with symptoms of hypokalemia and hypomagnesemia. Gitelman Syndrome:autosomal recessiveAutosomal recessiveAutosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal recessive diseases are only expressed when 2 copies of the recessive allele are inherited.Autosomal Recessive and Autosomal Dominant Inheritance disorder caused by one of several mutations in the genesGenesA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.DNA Types and Structure encoding sodiumSodiumA member of the alkali group of metals. It has the atomic symbol na, atomic number 11, and atomic weight 23.HyponatremiachlorideChlorideInorganic compounds derived from hydrochloric acid that contain the Cl- ion.Electrolytes and magnesiumMagnesiumA metallic element that has the atomic symbol mg, atomic number 12, and atomic weight 24. 31. It is important for the activity of many enzymes, especially those involved in oxidative phosphorylation.Electrolytes transporters in the thiazide-sensitive segments of the distal nephronNephronThe functional units of the kidney, consisting of the glomerulus and the attached tubule.Kidneys: Anatomy. Gitelman syndromeGitelman syndromeGitelman syndrome is a rare genetic autosomal recessive disorder that affects the sodium-chloride cotransporter in the distal convoluted tubule of the nephron and causes electrolyte abnormalities. The syndrome presents clinically with symptoms of hypokalemia and hypomagnesemia. Gitelman Syndrome is characterized by renal potassiumPotassiumAn element in the alkali group of metals with an atomic symbol k, atomic number 19, and atomic weight 39. 10. It is the chief cation in the intracellular fluid of muscle and other cells. Potassium ion is a strong electrolyte that plays a significant role in the regulation of fluid volume and maintenance of the water-electrolyte balance.Hyperkalemia loss, hypokalemiaHypokalemiaHypokalemia is defined as plasma potassium (K+) concentration < 3.5 mEq/L. Homeostatic mechanisms maintain plasma concentration between 3.5-5.2 mEq/L despite marked variation in dietary intake. Hypokalemia can be due to renal losses, GI losses, transcellular shifts, or poor dietary intake.Hypokalemia, metabolic alkalosisAlkalosisA pathological condition that removes acid or adds base to the body fluids.Respiratory Alkalosis, hypocalciuriaHypocalciuriaGitelman Syndrome, hypomagnesemiaHypomagnesemiaA nutritional condition produced by a deficiency of magnesium in the diet, characterized by anorexia, nausea, vomiting, lethargy, and weakness. Symptoms are paresthesias, muscle cramps, irritability, decreased attention span, and mental confusion, possibly requiring months to appear. Deficiency of body magnesium can exist even when serum values are normal. In addition, magnesium deficiency may be organ-selective, since certain tissues become deficient before others. Electrolytes, and hyperreninemic hyperaldosteronismHyperaldosteronismHyperaldosteronism is defined as the increased secretion of aldosterone from the zona glomerulosa of the adrenal cortex. Hyperaldosteronism may be primary (resulting from autonomous secretion), or secondary (resulting from physiological secretion due to stimulation of the RAAS). Classically, hyperaldosteronism presents with hypertension, hypokalemia, and metabolic alkalosis.Hyperaldosteronism with normal blood pressure. A key feature differentiating Gitelman syndromeGitelman syndromeGitelman syndrome is a rare genetic autosomal recessive disorder that affects the sodium-chloride cotransporter in the distal convoluted tubule of the nephron and causes electrolyte abnormalities. The syndrome presents clinically with symptoms of hypokalemia and hypomagnesemia. Gitelman Syndrome from Bartter syndrome is the urine calciumCalciumA basic element found in nearly all tissues. It is a member of the alkaline earth family of metals with the atomic symbol ca, atomic number 20, and atomic weight 40. Calcium is the most abundant mineral in the body and combines with phosphorus to form calcium phosphate in the bones and teeth. It is essential for the normal functioning of nerves and muscles and plays a role in blood coagulation (as factor IV) and in many enzymatic processes.Electrolytes level, which will typically be high-normal in BS and low in GS.
Diuretic abusewith loop diureticsDiureticsAgents that promote the excretion of urine through their effects on kidney function.Heart Failure and Chronic Coronary Syndrome Medication: target the TALTALRenal Sodium and Water Regulation and increase the excretion of sodiumSodiumA member of the alkali group of metals. It has the atomic symbol na, atomic number 11, and atomic weight 23.Hyponatremia, potassiumPotassiumAn element in the alkali group of metals with an atomic symbol k, atomic number 19, and atomic weight 39. 10. It is the chief cation in the intracellular fluid of muscle and other cells. Potassium ion is a strong electrolyte that plays a significant role in the regulation of fluid volume and maintenance of the water-electrolyte balance.Hyperkalemia, chlorideChlorideInorganic compounds derived from hydrochloric acid that contain the Cl- ion.Electrolytes, calciumCalciumA basic element found in nearly all tissues. It is a member of the alkaline earth family of metals with the atomic symbol ca, atomic number 20, and atomic weight 40. Calcium is the most abundant mineral in the body and combines with phosphorus to form calcium phosphate in the bones and teeth. It is essential for the normal functioning of nerves and muscles and plays a role in blood coagulation (as factor IV) and in many enzymatic processes.Electrolytes, magnesiumMagnesiumA metallic element that has the atomic symbol mg, atomic number 12, and atomic weight 24. 31. It is important for the activity of many enzymes, especially those involved in oxidative phosphorylation.Electrolytes, and water. HypokalemiaHypokalemiaHypokalemia is defined as plasma potassium (K+) concentration < 3.5 mEq/L. Homeostatic mechanisms maintain plasma concentration between 3.5-5.2 mEq/L despite marked variation in dietary intake. Hypokalemia can be due to renal losses, GI losses, transcellular shifts, or poor dietary intake.Hypokalemia is a common side effect and can be significant. Loop diureticsDiureticsAgents that promote the excretion of urine through their effects on kidney function.Heart Failure and Chronic Coronary Syndrome Medication are used mainly to treat edematous conditions, such as heart failureHeart FailureA heterogeneous condition in which the heart is unable to pump out sufficient blood to meet the metabolic need of the body. Heart failure can be caused by structural defects, functional abnormalities (ventricular dysfunction), or a sudden overload beyond its capacity. Chronic heart failure is more common than acute heart failure which results from sudden insult to cardiac function, such as myocardial infarction.Total Anomalous Pulmonary Venous Return (TAPVR) and cirrhosisCirrhosisCirrhosis is a late stage of hepatic parenchymal necrosis and scarring (fibrosis) most commonly due to hepatitis C infection and alcoholic liver disease. Patients may present with jaundice, ascites, and hepatosplenomegaly. Cirrhosis can also cause complications such as hepatic encephalopathy, portal hypertension, portal vein thrombosis, and hepatorenal syndrome. Cirrhosis, and they may also be used in the management of hypertensionHypertensionHypertension, or high blood pressure, is a common disease that manifests as elevated systemic arterial pressures. Hypertension is most often asymptomatic and is found incidentally as part of a routine physical examination or during triage for an unrelated medical encounter. Hypertension.
Cyclic vomitingVomitingThe forcible expulsion of the contents of the stomach through the mouth.Hypokalemia syndrome: condition characterized by recurrent, prolonged episodes of severe nauseaNauseaAn unpleasant sensation in the stomach usually accompanied by the urge to vomit. Common causes are early pregnancy, sea and motion sickness, emotional stress, intense pain, food poisoning, and various enteroviruses.Antiemetics and vomitingVomitingThe forcible expulsion of the contents of the stomach through the mouth.Hypokalemia. Metabolic alkalosisAlkalosisA pathological condition that removes acid or adds base to the body fluids.Respiratory Alkalosis and hypokalemiaHypokalemiaHypokalemia is defined as plasma potassium (K+) concentration < 3.5 mEq/L. Homeostatic mechanisms maintain plasma concentration between 3.5-5.2 mEq/L despite marked variation in dietary intake. Hypokalemia can be due to renal losses, GI losses, transcellular shifts, or poor dietary intake.Hypokalemia may result from GI losses. The cause is unknown and can begin at any age. Episodes of vomitingVomitingThe forcible expulsion of the contents of the stomach through the mouth.Hypokalemia may last hours or days with symptom-free intervals in between for weeks. Diagnosis is made clinically after ruling out other conditions. Management is aimed at controlling symptoms and avoiding triggers, as well as medications to prevent or relieve nauseaNauseaAn unpleasant sensation in the stomach usually accompanied by the urge to vomit. Common causes are early pregnancy, sea and motion sickness, emotional stress, intense pain, food poisoning, and various enteroviruses.Antiemetics.
BulimiaBulimiaEating an excess amount of food in a short period of time, as seen in the disorder of bulimia nervosa. It is caused by an abnormal craving for food, or insatiable hunger also known as ‘ox hunger’.Bulimia Nervosa nervosa: eating disorder characterized by recurrent episodes of binge eatingBinge eatingBinge eating is defined as consuming an amount of food in a specified amount of time (e.g., 1 hour) that greatly exceeds what most people would consume in that same amount of time.Binge Eating Disorder with inappropriate compensatory behaviorCompensatory behaviorBinge Eating Disorder, often including self-induced vomitingVomitingThe forcible expulsion of the contents of the stomach through the mouth.Hypokalemia and/or diuretic, laxativeLaxativeAgents that produce a soft formed stool, and relax and loosen the bowels, typically used over a protracted period, to relieve constipation.Hypokalemia, or thyroidThyroidThe thyroid gland is one of the largest endocrine glands in the human body. The thyroid gland is a highly vascular, brownish-red gland located in the visceral compartment of the anterior region of the neck.Thyroid Gland: Anatomy hormone abuse. BulimiaBulimiaEating an excess amount of food in a short period of time, as seen in the disorder of bulimia nervosa. It is caused by an abnormal craving for food, or insatiable hunger also known as ‘ox hunger’.Bulimia Nervosa nervosa frequently involves comorbid psychopathology. Treatment consists of psychotherapyPsychotherapyPsychotherapy is interpersonal treatment based on the understanding of psychological principles and mechanisms of mental disease. The treatment approach is often individualized, depending on the psychiatric condition(s) or circumstance. Psychotherapy and often psychopharmacologic agents.
Pyloric stenosisStenosisHypoplastic Left Heart Syndrome (HLHS): obstruction of outflow from the stomachStomachThe stomach is a muscular sac in the upper left portion of the abdomen that plays a critical role in digestion. The stomach develops from the foregut and connects the esophagus with the duodenum. Structurally, the stomach is C-shaped and forms a greater and lesser curvature and is divided grossly into regions: the cardia, fundus, body, and pylorus. Stomach: Anatomy due to hypertrophyHypertrophyGeneral increase in bulk of a part or organ due to cell enlargement and accumulation of fluids and secretions, not due to tumor formation, nor to an increase in the number of cells (hyperplasia).Cellular Adaptation and hyperplasiaHyperplasiaAn increase in the number of cells in a tissue or organ without tumor formation. It differs from hypertrophy, which is an increase in bulk without an increase in the number of cells.Cellular Adaptation of the pyloric sphincter muscle. Pyloric stenosisStenosisHypoplastic Left Heart Syndrome (HLHS) is the most common cause of GI obstruction in infants. Affected newborns typically present after the 3rd to 5th week of life with progressive nonbilious vomitingVomitingThe forcible expulsion of the contents of the stomach through the mouth.Hypokalemiaand a firm olive-like massMassThree-dimensional lesion that occupies a space within the breastImaging of the Breast in the epigastriumEpigastriumSurgical Anatomy of the Abdomen. Diagnosis is by ultrasonography, and management consists of fluid resuscitationResuscitationThe restoration to life or consciousness of one apparently dead. .Neonatal Respiratory Distress Syndrome, correction of electrolyte imbalances, and surgery.
HyperaldosteronismHyperaldosteronismHyperaldosteronism is defined as the increased secretion of aldosterone from the zona glomerulosa of the adrenal cortex. Hyperaldosteronism may be primary (resulting from autonomous secretion), or secondary (resulting from physiological secretion due to stimulation of the RAAS). Classically, hyperaldosteronism presents with hypertension, hypokalemia, and metabolic alkalosis.Hyperaldosteronism: defined as increased secretionSecretionCoagulation Studies of aldosteroneAldosteroneA hormone secreted by the adrenal cortex that regulates electrolyte and water balance by increasing the renal retention of sodium and the excretion of potassium.Hyperkalemia from the zona glomerulosaZona GlomerulosaThe narrow subcapsular outer zone of the adrenal cortex. This zone produces a series of enzymes that convert pregnenolone to aldosterone. The final steps involve three successive oxidations by cytochrome p-450 cyp11b2.Adrenal Glands: Anatomy of the adrenal cortexAdrenal CortexThe outer layer of the adrenal gland. It is derived from mesoderm and comprised of three zones (outer zona glomerulosa, middle zona fasciculata, and inner zona reticularis) with each producing various steroids preferentially, such as aldosterone; hydrocortisone; dehydroepiandrosterone; and androstenedione. Adrenal cortex function is regulated by pituitary adrenocorticotropin.Adrenal Glands: Anatomy. HyperaldosteronismHyperaldosteronismHyperaldosteronism is defined as the increased secretion of aldosterone from the zona glomerulosa of the adrenal cortex. Hyperaldosteronism may be primary (resulting from autonomous secretion), or secondary (resulting from physiological secretion due to stimulation of the RAAS). Classically, hyperaldosteronism presents with hypertension, hypokalemia, and metabolic alkalosis.Hyperaldosteronism may be primary or may be secondary to other causes. Classically, primary hyperaldosteronismPrimary hyperaldosteronismAutonomous (renin-independent) secretion of aldosterone.Hyperaldosteronism presents with hypertensionHypertensionHypertension, or high blood pressure, is a common disease that manifests as elevated systemic arterial pressures. Hypertension is most often asymptomatic and is found incidentally as part of a routine physical examination or during triage for an unrelated medical encounter. Hypertension, hypokalemiaHypokalemiaHypokalemia is defined as plasma potassium (K+) concentration < 3.5 mEq/L. Homeostatic mechanisms maintain plasma concentration between 3.5-5.2 mEq/L despite marked variation in dietary intake. Hypokalemia can be due to renal losses, GI losses, transcellular shifts, or poor dietary intake.Hypokalemia, and metabolic alkalosisAlkalosisA pathological condition that removes acid or adds base to the body fluids.Respiratory Alkalosis. Diagnosis is made by lab testing and imaging of the adrenal glandsAdrenal GlandsThe adrenal glands are a pair of retroperitoneal endocrine glands located above the kidneys. The outer parenchyma is called the adrenal cortex and has 3 distinct zones, each with its own secretory products. Beneath the cortex lies the adrenal medulla, which secretes catecholamines involved in the fight-or-flight response. Adrenal Glands: Anatomy. Management involves the use of aldosteroneAldosteroneA hormone secreted by the adrenal cortex that regulates electrolyte and water balance by increasing the renal retention of sodium and the excretion of potassium.HyperkalemiareceptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors antagonist medications and surgical excision of any aldosterone-secreting tumors.
Al Shibli, A., Narchi, H. (2015). Bartter and Gitelman syndromes: spectrum of clinical manifestations caused by different mutations.World Journal of Methodology, 5(2), 55–61. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4482822/