El síndrome de Bartter es un trastorno autosómico recesivo raro que afecta a los riñones y se presenta de forma prenatal con manifestaciones graves o potencialmente mortales o en la infancia o la adultez con un curso más leve, dependiendo del defecto genético. La enfermedad clínica es el resultado de una reabsorción renal defectuosa del cloruro de sodio en la rama ascendente gruesa del asa de Henle, donde normalmente se reabsorbe el 30% de la sal filtrada. El síndrome de Bartter se caracteriza por la pérdida de sal y la hipopotasemia y se presenta con anomalías electrolíticas y sus repercusiones, como vómitos y deshidratación. El diagnóstico se realiza mediante pruebas de laboratorio que muestran la típica alcalosis metabólica hipopotasémica e hipercalciuria. Otras anomalías de laboratorio son el aumento de la renina y la aldosterona en suero, aunque los pacientes evidencian clínicamente una presión arterial normal. El tratamiento se centra en la normalización de los niveles séricos de electrolitos. Los IECA y los ARA se utilizan para mejorar la hipopotasemia y limitar la proteinuria.
Last updated: Dec 15, 2025
El síndrome de Bartter es un trastorno genético raro (autosómico recesivo) que resulta de un defecto en la reabsorción de cloruro de sodio en la rama ascendente gruesa del asa de Henle, lo que conduce a la hipopotasemia y la alcalosis metabólica. El trastorno imita la ingesta prolongada de un diurético de asa.
En las células que recubren el asa de Henle:
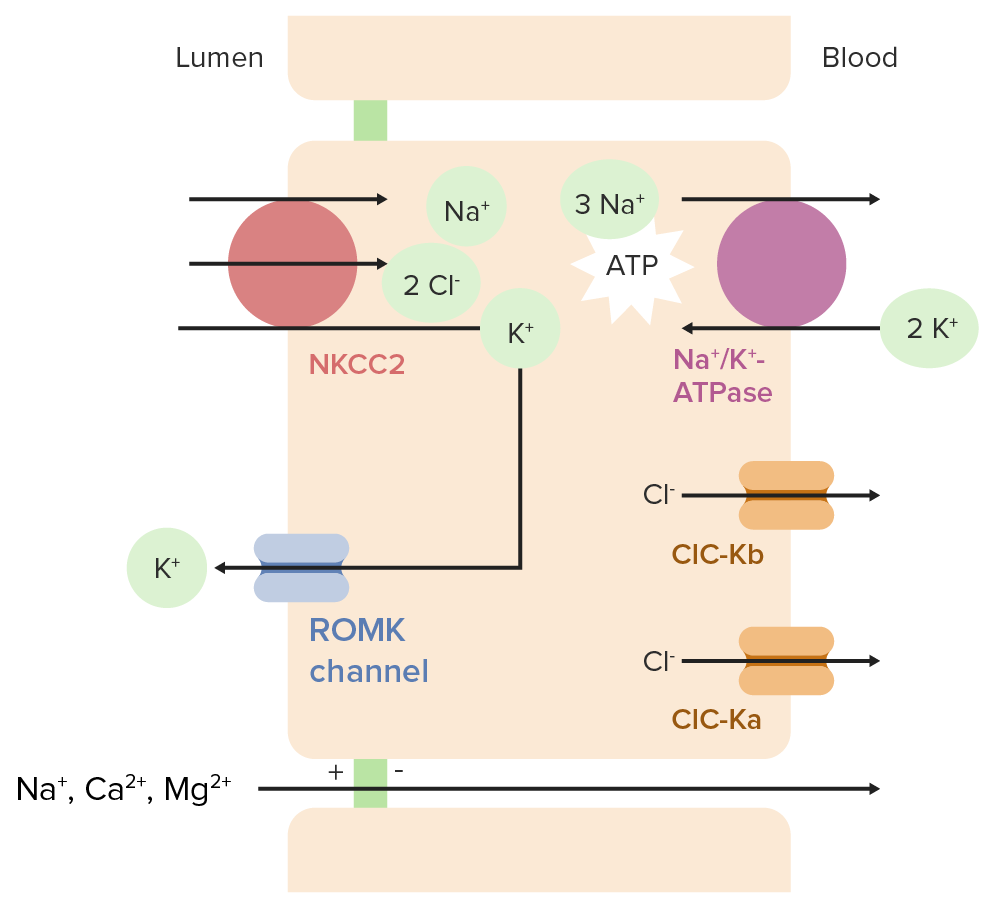
Transporte de iones en la rama gruesa ascendente del asa de Henle
ATPasa: adenosin trifosfatasa
ClC-Ka: canal de cloruro Ka
ClC-Kb: canal de cloruro Kb
NKCC2: cotransportador 2 de Na+/K+-Cl
ROMK: canales de potasio medulares externos renales
A causa de uno de varios defectos genéticos autosómicos recesivos, existe un trastorno tubular renal variable caracterizado por la pérdida de sal e hipopotasemia en todos los subtipos.
Existe una importante heterogeneidad genética en el síndrome de Bartter; puede ser el resultado de mutaciones homocigóticas o heterocigóticas mixtas en cualquiera de los genes que reducen la actividad de los transportadores de electrolitos en la rama ascendente gruesa del asa de Henle. Por lo tanto, la gravedad y la presentación clínica del síndrome de Bartter varían con cada tipo.
| Nombre | Tipo | Proteína defectuosa | Gravedad de la presentación |
|---|---|---|---|
| Síndrome de Bartter neonatal (síndrome de hiperprostaglandina E) | I | NKCC2 | Severo |
| Síndrome de Bartter neonatal | II | Canal ROMK | Severo |
| Síndrome de Bartter clásico | III | ClC-Kb | Leve |
| Síndrome de Bartter clásico con sordera neurosensorial (síndrome de hiperprostaglandina E) | IVa | Barttin (la subunidad β de ClC-Ka y ClC-Kb) | Severo |
| IVb | Mutaciones simultáneas en ClC-Ka y ClC-Kb | Severo | |
| Síndrome de Bartter con hipocalcemia (también llamado hipoparatiroidismo autosómico dominante) | V | CaSR | Leve |

Diferentes canalopatías en el síndrome de Bartter
ATPasa: adenosin trifosfatasa
ClC-Ka: canal de cloruro Ka
ClC-Kb: canal de cloruro Kb
NKCC2: cotransportador Na+/K+-Cl 2
ROMK: potasio medular externo renal
Las manifestaciones clínicas del síndrome de Bartter se deben principalmente a los desequilibrios electrolíticos y sus repercusiones. Los síntomas son mucho menos pronunciados en los heterocigotos. Los defectos tubulares en el transporte de iones producen un trastorno clínico que se parece al observado con la ingesta prolongada de un diurético de asa (e.g., furosemida).
Normalmente se observa en los tipos I, II, IVa y IVb. Los hallazgos más comunes son:
Niño con síndrome de Bartter que muestra una deshidratación grave antes del tratamiento
Imagen: “Bartter’s child before treatment showing severe dehydration” por Sampathkumar K et al. Licencia: CC BY 2.0Los hallazgos más comunes son:
El diagnóstico del síndrome de Bartter se realiza por los hallazgos de laboratorio tras la sospecha clínica que surge de los antecedentes y la exploración física.
Los defectos tubulares del síndrome de Bartter no pueden revertirse (excepto mediante un trasplante de riñón). El objetivo del tratamiento es disminuir los efectos de las prostaglandinas, la renina y la angiotensina elevadas en los tipos I, II y IV. En la forma adulta más leve, o síndrome de Bartter clásico, el objetivo principal es normalizar los niveles de potasio sérico.