A síndrome de Bartter é uma doença autossómica recessiva rara que afeta os rins e apresenta-se antenatalmente com manifestações graves ou com risco e vida na infância, ou na vida adulta com um curso maisMAISAndrogen Insensitivity Syndrome ligeiro, dependendo do defeito genético. As manifestações clínicas da doença resultam da reabsorção renal defeituosa do cloreto de sódio no segmento espesso do ramo ascendente da ansa de Henle, onde 30% do sal filtrado é normalmente reabsorvido. A síndrome de Bartter é caracterizada pela espoliação de sal e hipocalemia e apresenta distúrbios eletrolíticas e respetivas consequências, como vómitos e desidratação. O diagnóstico é feito por exames laboratoriais, que mostram a típica alcalose metabólica hipocalémica e hipercalciúria. Alterações laboratoriais adicionais incluem aumento da renina sérica e aldosterona, mas os pacientes têm pressão arterial normal. O tratamento centra-se na normalização dos níveis de eletrólitos séricos. Os inibidores da enzima conversora da angiotensina (IECA) e bloqueadores dos recetores de angiotensina são utilizados para melhorar a hipocalemia e limitar a proteinúria.
A síndrome de Bartter (SB) é uma doença genética rara (autossómica recessiva) que resulta de um defeito na reabsorção de cloreto de sódio no segmento espesso do ramo ascendente do laço de Henle, levando a hipocalemia e alcalose metabólica. Esta síndrome simula a toma de diuréticos de ansa a longo prazo.
Epidemiologia
Prevalência: 1 em 1 milhão de pessoas nos Estados Unidos
A síndrome de Bartter é menos comum que a síndrome de Gitelman (uma doença semelhante do túbulo renal, observada em 1-10 em 40.000 pessoas).
A prevalência de heterozigotos com uma das mutações genéticas que causam síndrome de Bartter é > 1% nos Estados Unidos e chega a 3% na Ásia.
Fisiopatologia e Classificação
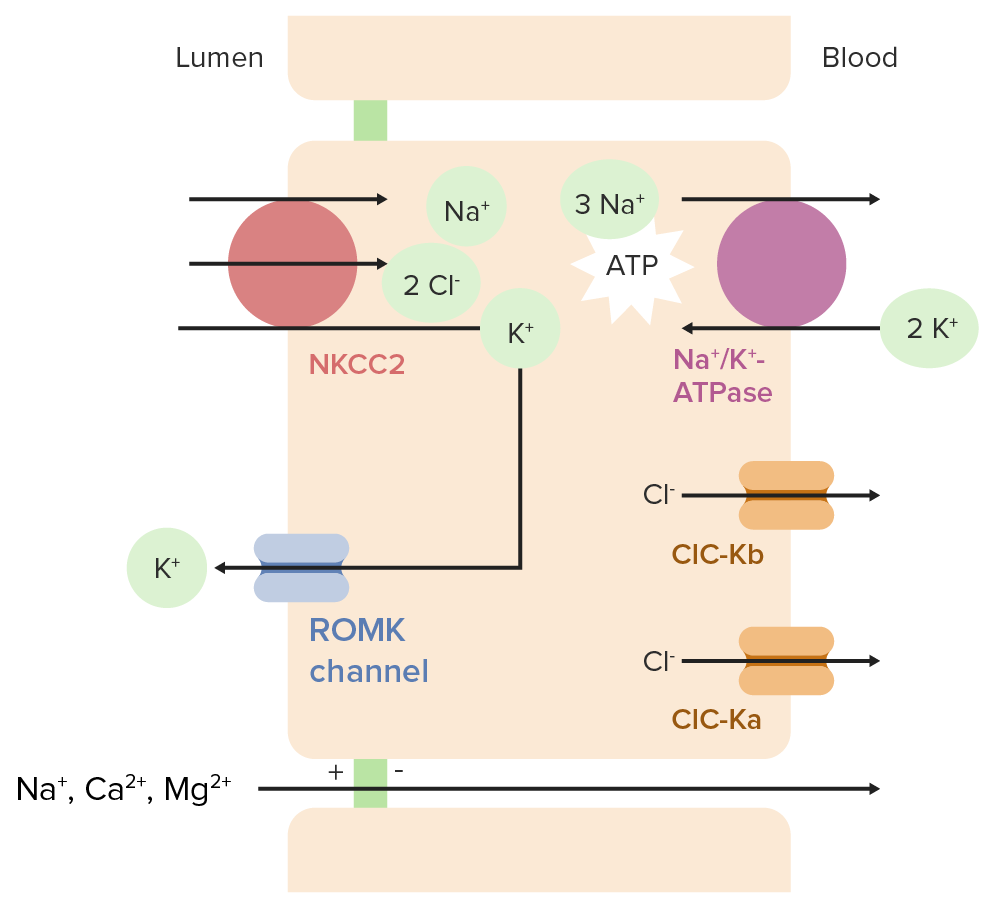
Fisiologia normal na ansa de Henle
Nas células que revestem a ansa de Henle:
A bomba de sódio/potássio-adenosina trifosfatase (Na+/K+-ATPase) na membrana basolateral leva 3 Na+ para fora da célula (para dentro do sangue) e 2 K+ para dentro da célula, resultando em:
↓ Concentração intracelular de Na+
↑ Concentração intracelular K+
O cotransportador Na+/K+-2Cl (também conhecido como NKCC2NKCC2Renal Potassium Regulation, da sigla em inglês) na membrana apical reabsorve 1 Na+, 1 K+, e 2 Cl– do lúmen tubular.
Eletroneutro (2 cargas positivas e 2 negativas movendo-se na mesma direção)
Impulsionado pela baixa concentração de Na+ dentro da célula
O ClC-Ka (canal de cloro Ka) é outro canal de cloro que se pensa ter uma função redundante
Para que o ClC-Kb e o ClC-Ka funcionem corretamente, é necessária uma pequena subunidade proteíca chamada barttin.
Nota: Estes mesmos canais de cloro são encontrados no ouvido, e as mutações nestes canais podem levar à surdez.
O K+ trazido para a célula através do NKCC2NKCC2Renal Potassium Regulation é reciclado de volta para o lúmen através dos canais de potássio renal externo medular (ROMK) → permite a reabsorção contínua de NaCl
Reabsorção de cálcio e magnésio:
O movimento de K+ carregado positivamente para o lúmen e Cl– carregado negativamente para fora do lúmen (e para dentro da corrente sanguínea) faz com que o lúmen fique maisMAISAndrogen Insensitivity Syndrome carregado positivamente do que o espaço peritubular.
Esta carga positiva impulsiona a reabsorção paracelular de Na+, Ca2+e Mg2+.
O transporte de iões no segmento espesso do ramo ascendente da ansa de Henle ATPase: adenosina trifosfatase ClC-Ka: canal de cloro Ka ClC-Kb: canal de cloro Kb NKCC2: cotransportador Na+/K+-Cl 2 ROMK: potássio renal medular externo
Imagem de Lecturio.
Fisiopatologia na síndrome de Bartter
Devido a um dos vários defeitos genéticos autossómicos recessivos, existe um distúrbio tubular renal variável, caracterizado pela espoliação de sal e hipocalemia em todos os subtipos.
A síndrome de Bartter é caracterizada pelo comprometimento do NKCC2NKCC2Renal Potassium Regulation, que se encontra na porção espessa do ramo ascendente da ansa de Henle.
A deficiência deste transportador vai ↓ reabsorção na porção espessa do ramo ascendente da ansa de Henle de:
Sódio
Potássio
Cloro
↓ Reabsorção de iões → leva ao ↑ da entrega distal destes iões
Estes iões permanecem no lúmen tubular enquanto a urina se desloca distalmente.
↑ Entrega distal de Na+ → ↑ o gradiente de eletronegatividade através da membrana luminal → ↑ excreção de K+ → hipocalemia
↑ Entrega distal de K+ → ↑ troca de K+ por H+ no tubo coletor → ↑ excreção de H+ → alcalose metabólica
↑ Na+ na urina → ↑ água livre na urina, resultando em:
Incapacidade de concentrar a urina
Depleção de volume → causa a ativação do SRAA e leva ao hiperaldosteronismo secundário
A estimulação a longo prazo da SRAA causa hiperplasia do aparelho justaglomerular e aumento dos níveis de renina.
↑ Libertação renal de prostaglandina E2
↑ Fluxo de sangue renal e taxa de filtração glomerular
↑ Secreção de Renina
↑ Na+ e excreção de água livre
Também ↑da perda urinária de cálcio e magnésio.
↓ Reabsorção de NaCl → ↓ Reabsorção de CaCACondylomata acuminata are a clinical manifestation of genital HPV infection. Condylomata acuminata are described as raised, pearly, flesh-colored, papular, cauliflower-like lesions seen in the anogenital region that may cause itching, pain, or bleeding.Condylomata Acuminata (Genital Warts)2+ e Mg+
A espoliação de cálcio na urina pode levar a nefrocalcinose.
A ativação de mutações no recetor sensor de cálcio (CaSR) na membrana basolateral também pode prejudicar o transporte de NaCl → provoca uma forma ligeira de SB
Também leva a um “resetting” para baixo do nível de cálcio sérico normal → resulta na ↓ hormona paratiroideia (PTH) e hipocalcemia
Classificação por mutações
Existe uma heterogeneidade genética significativa na SB; pode resultar de mutações homozigotas ou heterozigotas mistas em qualquer um dos genesGenesA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.DNA Types and Structure que reduzem a atividade dos transportadores de eletrólitos na porção espessa do ramo ascendente da ansa de Henle. Assim, a gravidade e a apresentação clínica da SB variam entre cada tipo.
Note-se que a hipocalcemia autossómica dominante, uma condição associada a uma mutação ativadora do geneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of GeneticsCASR (que afeta o recetor sensível ao cálcio), era anteriormente classificada como síndrome de Bartter tipo V. Alguns casos podem apresentar perda de potássio e alcalose metabólica, semelhante à síndrome de Bartter.
A síndrome de Bartter tipo V é uma síndrome de Bartter pré-natal transitória, uma forma grave, mas transitória. Está associada a mutações no MAGED2, um geneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics no cromossoma X que codifica o antígeno D2 associado ao melanomaMelanomaMelanoma is a malignant tumor arising from melanocytes, the melanin-producing cells of the epidermis. These tumors are most common in fair-skinned individuals with a history of excessive sun exposure and sunburns. Melanoma (MAGE-D2). Isso afeta a reabsorção renal fetal de sal e a apresentação inclui polidrâmnio, prematuridade, hiponatremia, alcalose metabólica e poliúria.
Tabela: Resumo de 5 subtipos da síndrome de Bartter, dependendo do geneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics envolvido e do transportador afetado
Nome
Tipo
Proteína com defeito
Gravidade da apresentação clínica
Síndrome de Bartter Neonatal (síndrome de hiperprostaglandina E)
Síndrome de Bartter clássica com surdez neurossensorial (síndrome de hiperprostaglandina E)
IVa
Barttin (a subunidade β de ClC-Ka e ClC-Kb)
Grave
IVb
Mutações simultâneas no ClC-Ka e no ClC-Kb
Grave
Síndrome de Bartter com hipocalcemia (também chamado hipoparatiroidismo autossómico dominante)
V
CaSR
Ligeira
Diferentes canalopatias na síndrome de Bartter ATPase: adenosina trifosfatase ClC-Ka: canal de cloro Ka ClC-Kb: canal de cloro Kb NKCC2: cotransportador Na+/K+-Cl 2 ROMK: potássio renal medular externo
As manifestações clínicas da SB são devidas principalmente a desequilíbrios eletrolíticos e respetivas consequências. Os sintomas são muito menos pronunciados nos heterozigotos. Os defeitos tubulares no transporte de iões produzem um distúrbio clínico semelhante ao observado com a toma de um diurético de ansa (por exemplo, furosemida) a longo prazo.
Hipofosfatemia em alguns pacientes com hiperparatiroidismo secundário
Nível sérico normal ou ligeiramente diminuído de magnésio
Vómitos
Poliúria e polidipsia devido à diminuição da capacidade de concentração urinária
Cãibras abdominais
Desidratação/hipovolemia
Hipotensão
Obstipação
Fraqueza muscular
Disfunção renal (manifestação tardia):
Proteinúria
↓ TFG
Diagnóstico
O diagnóstico de SB é realizado por achados laboratoriais após a suspeita clínica da história clínica e exame físico.
História clínica e exame objetivo
Avaliar para vómitos sub-reptícios; os achados podem incluir:
Cicatrizes na mão por inserção na boca
Erosão dentária
Parotidite
Excluir o uso de diuréticos não prescritos.
Perguntar sobre história familiar de nefrocalcinose.
Observar características faciais.
Hipotensão (o hiperaldosteronismo primário apresentar-se-á normalmente com hipertensão).
Estudo laboratorial
Electrólitos séricos:
Hipocalemia inexplicada (uma característica chave no diagnóstico da SB)
Alcalose metabólica
Hipomagnesemia
Hipofosfatemia
Electrólitos urinários:
↑ Cloro urinário (diferencia a SB dos vómitos sub-reptícios, que terão um ↓ Cl– urinário)
↑ Cálcio urinário (diferencia a SB da síndrome de Gitelman, que terá uma ↓ do CaCACondylomata acuminata are a clinical manifestation of genital HPV infection. Condylomata acuminata are described as raised, pearly, flesh-colored, papular, cauliflower-like lesions seen in the anogenital region that may cause itching, pain, or bleeding.Condylomata Acuminata (Genital Warts)2+ urinário)
↑ Sódio urinário
↑ Potássio urinário
Alterações adicionais do estudo laboratorial:
↑ renina sérica
↑ aldosterona sérica
↑ Prostaglandina E2
↑ PTH
Podem ser utilizados testesTestesGonadal Hormones genéticos para procurar por mutações específicas.
Os níveis de cloro do fluido amniótico podem estar elevados.
O nível de alfa-fetoproteína está baixo na SB pré-natal.
Tratamento e Complicações
Os defeitos tubulares na SB não podemser corrigidos (exceto por transplante renal). O objetivo do tratamento é diminuir os efeitos da elevação dos níveis das prostaglandinas, renina e angiotensina nos tipos I, II e IV. Na forma adulta maisMAISAndrogen Insensitivity Syndrome ligeira, ou SB clássica, o objetivo principal é normalizar os níveis séricos de potássio.
Síndrome de Bartter pré-natal com polidrâmnio grave
AINEs para antagonizar os efeitos da ↑ das prostaglandinas
Evitar os AINE após as 32 semanas de gestação (pode causar o encerramento prematuro do canal arterial).
Se forem utilizados AINEs, deve ser repetida uma avaliação ecografia para averiguar o desenvolvimento de regurgitação tricúspide
Considerar amniocentese intermitente no 3º trimestre para tratar polidrâmnios graves através da drenagem excessiva de líquido amniótico.
Síndrome de Bartter Neonatal tipos I, II, e IV
Pode ser necessária uma infusão salina intravenosa para tratar a desidratação.
Será provavelmente necessária a suplementação oral de potássio.
Síndrome de Bartter infantil ou do adulto tipo III
Suplementação oral de eletrólitos:
Para corrigir desequilíbrios de fluidos e eletrólitos
Antagonizar os efeitos do aumento das prostaglandinas
Exemplos: indometacina, celecoxibCelecoxibA pyrazole derivative and selective cyclooxygenase 2 inhibitor that is used to treat symptoms associated with rheumatoid arthritis; osteoarthritis and juvenile arthritis, as well as the management of acute pain.Nonsteroidal Antiinflammatory Drugs (NSAIDs)
Diuréticos poupadores de potássio:
Bloqueiam a troca tubulo-distal sódio-potássio → pode ↑ K+ sérico e reverter a alcalose metabólica
Exemplos: amilorida, espironolactona
Inibidores da ECA e bloqueadores dos recetores de angiotensina:
Diminuem os níveis elevados de angiotensina II e aldosterona
Limitam a proteinúria
Aumentam o potássio sérico
Transplante renal:
Para raros doentes com doença renal em fase terminal e/ou nefrocalcinose
As anomalias tubulares resolvem após o transplante renal, sem recorrência.
Complicações
Arritmias cardíacas e morte súbita cardíaca devido a desequilíbrios eletrolíticos
Síndrome de Gitelman: distúrbio autossómico recessivo causado por uma das várias mutações nos genesGenesA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.DNA Types and Structure que codificam os transportadores de cloreto de sódio e magnésio nos segmentos sensíveis à tiazida do nefrónio distal. A síndrome de Gitelman é caracterizada por perda renal de potássio, hipocalcemia, alcalose metabólica, hipocalciúria, hipomagnesemia e hiperaldosteronismo hiperreninémico com pressão arterial normal. Uma característica chave que diferencia a síndrome de Gitelman da síndrome de Bartter é o nível de cálcio urinário, que normalmente será normal-alto na SB e baixo na SG.
Abuso de diuréticoscom diuréticos de ansa: têm como alvo a porção espessa do ramo ascendente da ansa de Henle e aumentam a excreção de sódio, potássio, cloreto, cálcio, magnésio e água. A hipocalemia é um efeito adverso comum e pode ser significativa. Os diuréticos da ansa são utilizados principalmente no tratamento de patologias edematosas, como insuficiência cardíaca e cirrose. Também podem ser usados no tratamento da hipertensão.
Síndrome de vómitos cíclicos: condição caracterizada por episódios recorrentes e prolongados de náuseas graves e vómitos. A alcalose metabólica e a hipocalemia podem resultar de perdas gastrointestinais. A causa é desconhecida e pode começar em qualquer idade. Os episódios de vómitos podem durar horas ou dias, com intervalos assintomáticos durante semanas. O diagnóstico é clínico, após exclusão de outras condições. O tratamento visa controlar os sintomas e evitar os desencadeantes, para além do uso de medicamentos para prevenir ou aliviar as náuseas.
BulimiaBulimiaEating an excess amount of food in a short period of time, as seen in the disorder of bulimia nervosa. It is caused by an abnormal craving for food, or insatiable hunger also known as ‘ox hunger’.Bulimia Nervosa nervosa: distúrbio alimentar caracterizado por episódios recorrentes de compulsão alimentar com comportamento compensatório inadequado, muitas vezes incluindo vómitos autoinduzidos e/ou abuso de diuréticos, laxantes ou hormonas tiroideias. A bulimiaBulimiaEating an excess amount of food in a short period of time, as seen in the disorder of bulimia nervosa. It is caused by an abnormal craving for food, or insatiable hunger also known as ‘ox hunger’.Bulimia Nervosa nervosa está frequentemente associada a psicopatologia. O tratamento consiste em psicoterapia e muitas vezes em agentes psicofarmacológicos.
Estenose pilórica: obstrução da saída do estômago, devido à hipertrofia e hiperplasia do esfíncter pilórico. A estenose pilórica é a causa maisMAISAndrogen Insensitivity Syndrome comum de obstrução gastrointestinal em crianças. Os recém-nascidos afetados apresentam tipicamente vómitos progressivos e não-biliares após a 3ª a 5ª semana de vida.e uma massa firme com forma semelhante a azeitona no epigástrio. O diagnóstico é feito por ecografia, e o tratamento consiste em fluidoterapia, correção de desequilíbrios eletrolíticos e cirurgia.
Hiperaldosteronismo: definido como aumento da secreção de aldosterona a partir da zona glomerulosaZona GlomerulosaThe narrow subcapsular outer zone of the adrenal cortex. This zone produces a series of enzymes that convert pregnenolone to aldosterone. The final steps involve three successive oxidations by cytochrome p-450 cyp11b2.Adrenal Glands: Anatomy do córtex adrenal. O hiperaldosteronismo pode ser primário ou secundário a outras causas. Classicamente, o hiperaldosteronismo primário apresenta hipertensão, hipocalemia e alcalose metabólica. O diagnóstico é feito através de testesTestesGonadal Hormones laboratoriais e exames de imagem das glândulas suprarrenais. O tratamento envolve o uso de medicamentos antagonistas dos recetores de aldosterona e a excisão cirúrgica de quaisquer tumores secretores de aldosterona.
Al Shibli, A., Narchi, H. (2015). Bartter and Gitelman syndromes: spectrum of clinical manifestations caused by different mutations.World Journal of Methodology 5(2):55–61.
A Lecturio Medical complementa o teu estudo através de métodos de ensino baseados em evidência, vídeos de palestras, perguntas e muito mais – tudo combinado num só lugar e fácil de usar.
User Reviews
Details
×
Obtenha Premium para testar os seus conhecimentos
Lecturio Premium dá-lhe acesso total a todos os conteúdos e características
Obtenha Premium para ver todos os vídeos
Verifique agora o seu e-mail para obter um teste gratuito.
Crie uma conta gratuita para testar os seus conhecimentos
Lecturio Premium dá-lhe acesso total a todos os conteúdos e características - incluindo o Qbank de Lecturio com perguntas actualizadas ao estilo do board-.