El síndrome de Gitelman es un raro trastorno genético autosómico recesivo que afecta al AL Amyloidosis cotransportador de sodio-cloruro en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el túbulo contorneado distal de la nefrona y provoca anomalías electrolíticas. El síndrome se presenta clínicamente con síntomas de hipopotasemia e hipomagnesemia. El diagnóstico se basa en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la presentación clínica y las pruebas de laboratorio (que muestran hipocalemia, hipomagnesemia, alcalosis metabólica e hipocalciuria), y se confirma con pruebas genéticas. El pilar del tratamiento es la administración de suplementos de electrolitos. El pronóstico es bueno, pero hay que vigilar la hipopotasemia para evitar arritmias cardíacas y posibles paros cardíacos.
Last updated: Dec 15, 2025
El síndrome de Gitelman es un raro trastorno genético autosómico recesivo que afecta al AL Amyloidosis cotransportador Na+–Cl– sensible a las tiazidas en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el túbulo contorneado distal, lo que provoca una tubulopatía por pérdida de sal.
El túbulo contorneado distal es la porción más pequeña del sistema de conductos en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum una nefrona. Mide unos 5 mm de tamaño y se origina en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la mácula densa. Las características del túbulo contorneado distal son las siguientes:
El síndrome de Gitelman conduce a la pérdida de la función del cotransportador Na+–Cl– → anomalías electrolíticas debido a la interferencia con el funcionamiento normal del túbulo contorneado distal
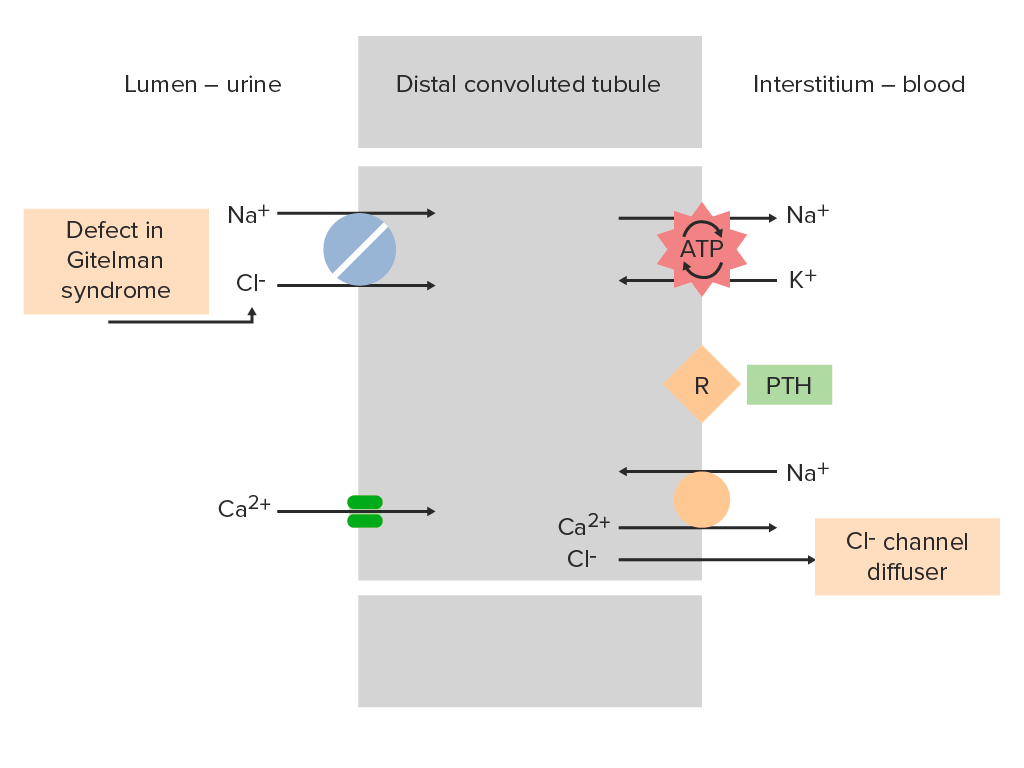
Cotransportador de sodio-cloruro en el túbulo contorneado distal de una nefrona:
El cotransportador de sodio-cloruro normalmente ayuda a la reabsorción de Na+ y Cl- desde el lumen del túbulo. En el síndrome de Gitelman, la inactivación del cotransportador de sodio-cloruro impide esta absorción, aumentando la entrega de electrolitos al conducto colector. Este fenómeno es similar al mecanismo de acción de los diuréticos tiazídicos.
PTH: hormona paratiroidea
R: receptor de la hormona paratiroidea
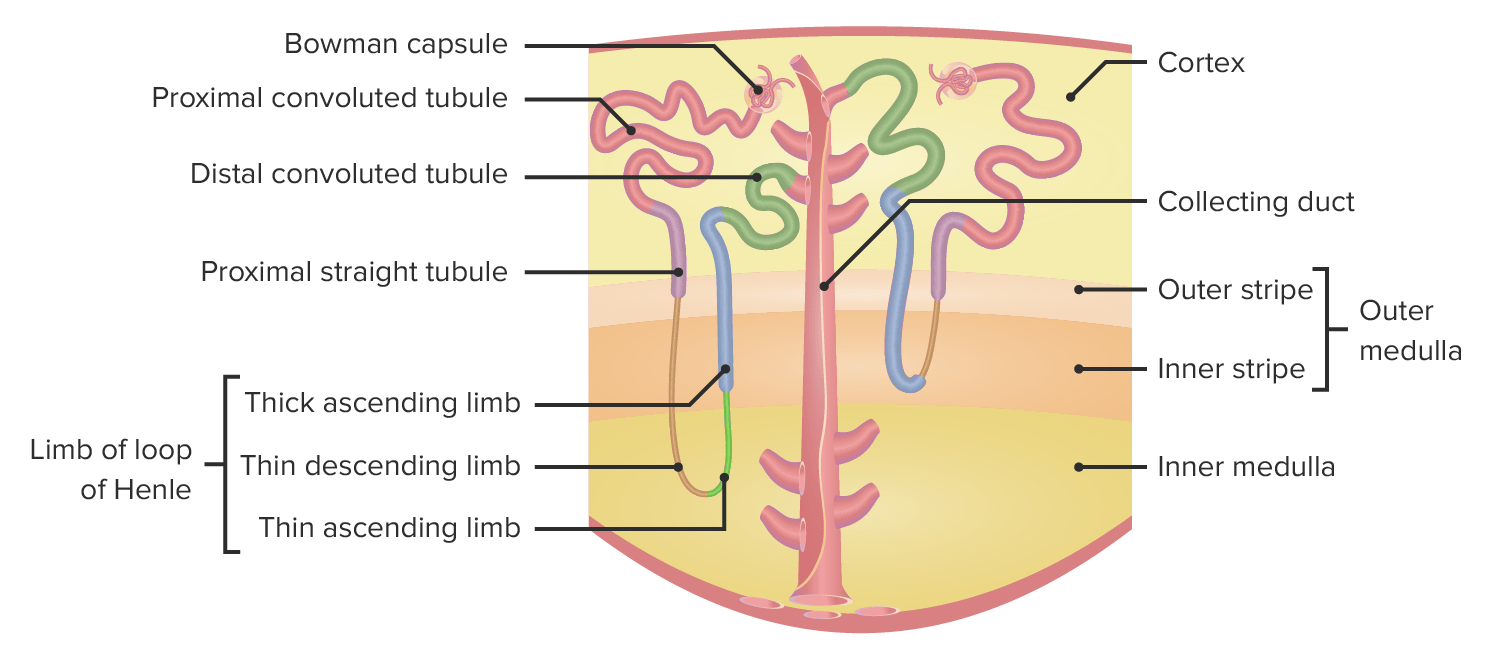
Anatomía de una nefrona:
El verde representa el túbulo contorneado distal, que está afectado en los individuos con síndrome de Gitelman.
Las personas con síndrome de Gitelman tienen síntomas de leves a moderados sin limitación en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la actividad diaria. Estos individuos se presentan después de la primera década de vida en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la adolescencia o en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la adultez temprana (rara vez en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la infancia).
La presentación clínica clásica es la tríada de:
Otros signos y síntomas son:
Es necesaria una evaluación detallada de síndrome de Gitelman cuando un individuo presenta hipopotasemia inexplicable, alcalosis metabólica y presión arterial normal o baja. Debido a su rara aparición en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum comparación con otros trastornos renales con síntomas similares, se deben descartar otras causas.
Las pruebas genéticas son altamente específicas y sensibles, y la mayoría de las personas afectadas muestran mutaciones en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum 2 genes Genes A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. DNA Types and Structure particulares:
Los LOS Neisseria objetivos del tratamiento son minimizar los LOS Neisseria efectos del agotamiento del volumen extracelular y corregir las deficiencias de electrolitos.