The term "disorders of sexual development" refers to a group of conditions characterized by atypical sexual development in an individual, which may involve abnormalities in the structure and/or function of the internal reproductive organs and/or external genitalia. Typical sexSexThe totality of characteristics of reproductive structure, functions, phenotype, and genotype, differentiating the male from the female organism.Gender Dysphoria development starts with the chromosomal sexChromosomal sexSex Determination (e.g., 46,XY or 46,XX), which determines the sexual differentiation of the gonadsGonadsThe gamete-producing glands, ovary or testis.Hormones: Overview and Types (e.g., testesTestesGonadal Hormones or ovariesOvariesOvaries are the paired gonads of the female reproductive system that contain haploid gametes known as oocytes. The ovaries are located intraperitoneally in the pelvis, just posterior to the broad ligament, and are connected to the pelvic sidewall and to the uterus by ligaments. These organs function to secrete hormones (estrogen and progesterone) and to produce the female germ cells (oocytes).Ovaries: Anatomy), which secrete hormonesHormonesHormones are messenger molecules that are synthesized in one part of the body and move through the bloodstream to exert specific regulatory effects on another part of the body. Hormones play critical roles in coordinating cellular activities throughout the body in response to the constant changes in both the internal and external environments. Hormones: Overview and Types that determine the phenotypePhenotypeThe complete genetic complement contained in the DNA of a set of chromosomes in a human. The length of the human genome is about 3 billion base pairs.Basic Terms of Genetics (e.g., male or female). Most disorders of sexual development are due to abnormalities in specific chromosomesChromosomesIn a prokaryotic cell or in the nucleus of a eukaryotic cell, a structure consisting of or containing DNA which carries the genetic information essential to the cell.DNA Types and Structure, enzymesEnzymesEnzymes are complex protein biocatalysts that accelerate chemical reactions without being consumed by them. Due to the body's constant metabolic needs, the absence of enzymes would make life unsustainable, as reactions would occur too slowly without these molecules. Basics of Enzymes, or receptorsReceptorsReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors. Diagnosis typically involves analyzing the karyotypeKaryotypeThe full set of chromosomes presented as a systematized array of metaphase chromosomes from a photomicrograph of a single cell nucleus arranged in pairs in descending order of size and according to the position of the centromere.Congenital Malformations of the Female Reproductive System and specific hormone levels. Management can be complex, and often includes psychotherapyPsychotherapyPsychotherapy is interpersonal treatment based on the understanding of psychological principles and mechanisms of mental disease. The treatment approach is often individualized, depending on the psychiatric condition(s) or circumstance. Psychotherapy, hormone replacement therapyHormone Replacement TherapyHormone replacement therapy (HRT) is used to treat symptoms associated with female menopause and in combination to suppress ovulation. Risks and side effects include uterine bleeding, predisposition to cancer, breast tenderness, hyperpigmentation, migraine headaches, hypertension, bloating, and mood changes.Noncontraceptive Estrogen and Progestins, and/or surgery.
Disorders of sexual development are a group of conditions characterized by atypical sexual development in an individual, involving abnormalities in the structure and/or function of both the internal reproductive organs and/or external genitalia.
Overview of typical sexSexThe totality of characteristics of reproductive structure, functions, phenotype, and genotype, differentiating the male from the female organism.Gender Dysphoria development
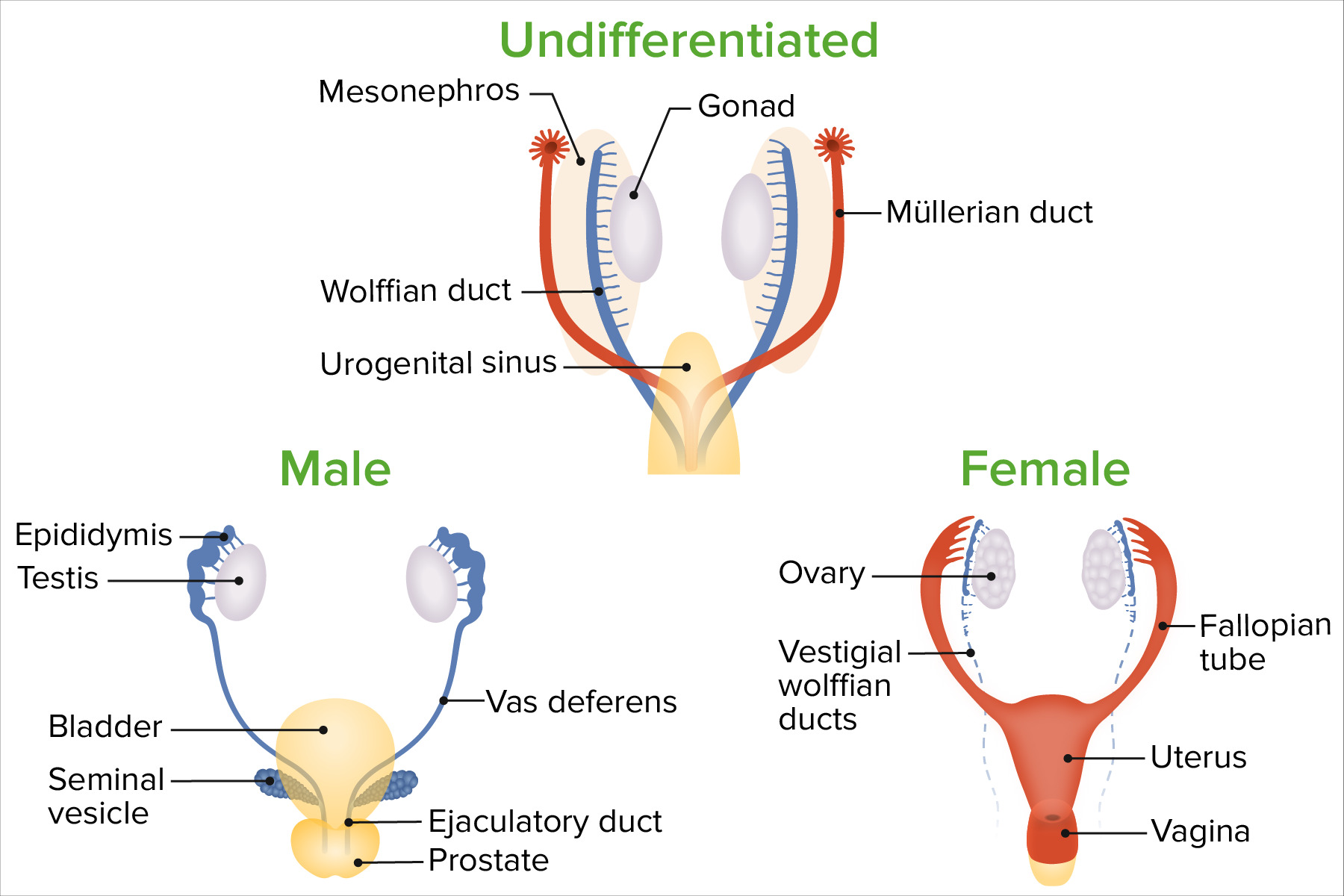
Up until 6 weeks of gestation, sexSexThe totality of characteristics of reproductive structure, functions, phenotype, and genotype, differentiating the male from the female organism.Gender Dysphoria development is identical and nonbinary. The developing structures include:
The genesGenesA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.DNA Types and Structure present at fertilizationFertilizationTo undergo fertilization, the sperm enters the uterus, travels towards the ampulla of the fallopian tube, and encounters the oocyte. The zona pellucida (the outer layer of the oocyte) deteriorates along with the zygote, which travels towards the uterus and eventually forms a blastocyst, allowing for implantation to occur. Fertilization and First Week will determine how the developing bipotent gonadsGonadsThe gamete-producing glands, ovary or testis.Hormones: Overview and Types differentiate (e.g., into a testis or an ovary).
The developing gonadsGonadsThe gamete-producing glands, ovary or testis.Hormones: Overview and Types will then secrete hormonesHormonesHormones are messenger molecules that are synthesized in one part of the body and move through the bloodstream to exert specific regulatory effects on another part of the body. Hormones play critical roles in coordinating cellular activities throughout the body in response to the constant changes in both the internal and external environments. Hormones: Overview and Types:
The presence and/or absence of specific hormonesHormonesHormones are messenger molecules that are synthesized in one part of the body and move through the bloodstream to exert specific regulatory effects on another part of the body. Hormones play critical roles in coordinating cellular activities throughout the body in response to the constant changes in both the internal and external environments. Hormones: Overview and Types will determine how the remaining structures differentiate.
In general, female organs and structures are the “default” phenotypePhenotypeThe complete genetic complement contained in the DNA of a set of chromosomes in a human. The length of the human genome is about 3 billion base pairs.Basic Terms of Genetics if specific genesGenesA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.DNA Types and Structure and hormonesHormonesHormones are messenger molecules that are synthesized in one part of the body and move through the bloodstream to exert specific regulatory effects on another part of the body. Hormones play critical roles in coordinating cellular activities throughout the body in response to the constant changes in both the internal and external environments. Hormones: Overview and Types are not present to stimulate male differentiation.
SexSexThe totality of characteristics of reproductive structure, functions, phenotype, and genotype, differentiating the male from the female organism.Gender Dysphoria organs and characteristics
GonadsGonadsThe gamete-producing glands, ovary or testis.Hormones: Overview and Types develop based on the karyotypeKaryotypeThe full set of chromosomes presented as a systematized array of metaphase chromosomes from a photomicrograph of a single cell nucleus arranged in pairs in descending order of size and according to the position of the centromere.Congenital Malformations of the Female Reproductive System/genesGenesA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.DNA Types and Structure that are present.
Develop when sex-determining region of the Y chromosomeY chromosomeThe male sex chromosome, being the differential sex chromosome carried by half the male gametes and none of the female gametes in humans and in some other male-heterogametic species in which the homologue of the X chromosome has been retained.Basic Terms of Genetics (SRY geneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics) is present
SecretetestosteroneTestosteroneA potent androgenic steroid and major product secreted by the leydig cells of the testis. Its production is stimulated by luteinizing hormone from the pituitary gland. In turn, testosterone exerts feedback control of the pituitary LH and FSH secretion. Depending on the tissues, testosterone can be further converted to dihydrotestosterone or estradiol.Androgens and Antiandrogensand anti-Müllerian hormone (AMHAMHA glycoprotein that causes regression of mullerian ducts. It is produced by sertoli cells of the testes. In the absence of this hormone, the mullerian ducts develop into structures of the female reproductive tract. In males, defects of this hormone result in persistent mullerian duct, a form of male pseudohermaphroditism.Primary Amenorrhea)
OvariesOvariesOvaries are the paired gonads of the female reproductive system that contain haploid gametes known as oocytes. The ovaries are located intraperitoneally in the pelvis, just posterior to the broad ligament, and are connected to the pelvic sidewall and to the uterus by ligaments. These organs function to secrete hormones (estrogen and progesterone) and to produce the female germ cells (oocytes).Ovaries: Anatomy develop when the X chromosomeX chromosomeThe female sex chromosome, being the differential sex chromosome carried by half the male gametes and all female gametes in human and other male-heterogametic species.Basic Terms of Genetics is present and/or the SRY is absent.
Ovotestis: a gonad containing both ovarian and testicular tissue found in patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship with true hermaphroditismTrue hermaphroditismTrue hermaphroditism, or ovotesticular disorder of sexual development (ODSD), is characterized by the presence of an ovotesticular gonad that contains both ovarian and testicular elements. Individuals are usually born with ambiguous genitalia, but the diagnosis is rarely confirmed before puberty. The most common karyotype is 46,XX, and less often, 46,XY can be identified.Ovotesticular Disorder of Sexual Development (ODSD)
Wolffian structures differentiate from the Wolffian (mesonephric) ducts in the presence of testosteroneTestosteroneA potent androgenic steroid and major product secreted by the leydig cells of the testis. Its production is stimulated by luteinizing hormone from the pituitary gland. In turn, testosterone exerts feedback control of the pituitary LH and FSH secretion. Depending on the tissues, testosterone can be further converted to dihydrotestosterone or estradiol.Androgens and Antiandrogens:
EpididymisEpididymisThe convoluted cordlike structure attached to the posterior of the testis. Epididymis consists of the head (caput), the body (corpus), and the tail (cauda). A network of ducts leaving the testis joins into a common epididymal tubule proper which provides the transport, storage, and maturation of spermatozoa.Testicles: Anatomy
Vas deferensVas DeferensThe excretory duct of the testes that carries spermatozoa. It rises from the scrotum and joins the seminal vesicles to form the ejaculatory duct.Testicles: Anatomy
Ejaculatory ductsEjaculatory DuctsPaired ducts in the human male through which semen is ejaculated into the urethra.
Müllerian structures differentiate from the Müllerian (paramesonephric) ducts when AMHAMHA glycoprotein that causes regression of mullerian ducts. It is produced by sertoli cells of the testes. In the absence of this hormone, the mullerian ducts develop into structures of the female reproductive tract. In males, defects of this hormone result in persistent mullerian duct, a form of male pseudohermaphroditism.Primary Amenorrhea isabsent:
UterusUterusThe uterus, cervix, and fallopian tubes are part of the internal female reproductive system. The uterus has a thick wall made of smooth muscle (the myometrium) and an inner mucosal layer (the endometrium). The most inferior portion of the uterus is the cervix, which connects the uterine cavity to the vagina.Uterus, Cervix, and Fallopian Tubes: Anatomy
Fallopian tubesFallopian tubesThe uterus, cervix, and fallopian tubes are part of the internal female reproductive system. The fallopian tubes receive an ovum after ovulation and help move it and/or a fertilized embryo toward the uterus via ciliated cells lining the tubes and peristaltic movements of its smooth muscle. Uterus, Cervix, and Fallopian Tubes: Anatomy
Upper ⅓ of the vaginaVaginaThe vagina is the female genital canal, extending from the vulva externally to the cervix uteri internally. The structures have sexual, reproductive, and urinary functions and a rich blood supply, mainly arising from the internal iliac artery.Vagina, Vulva, and Pelvic Floor: Anatomy
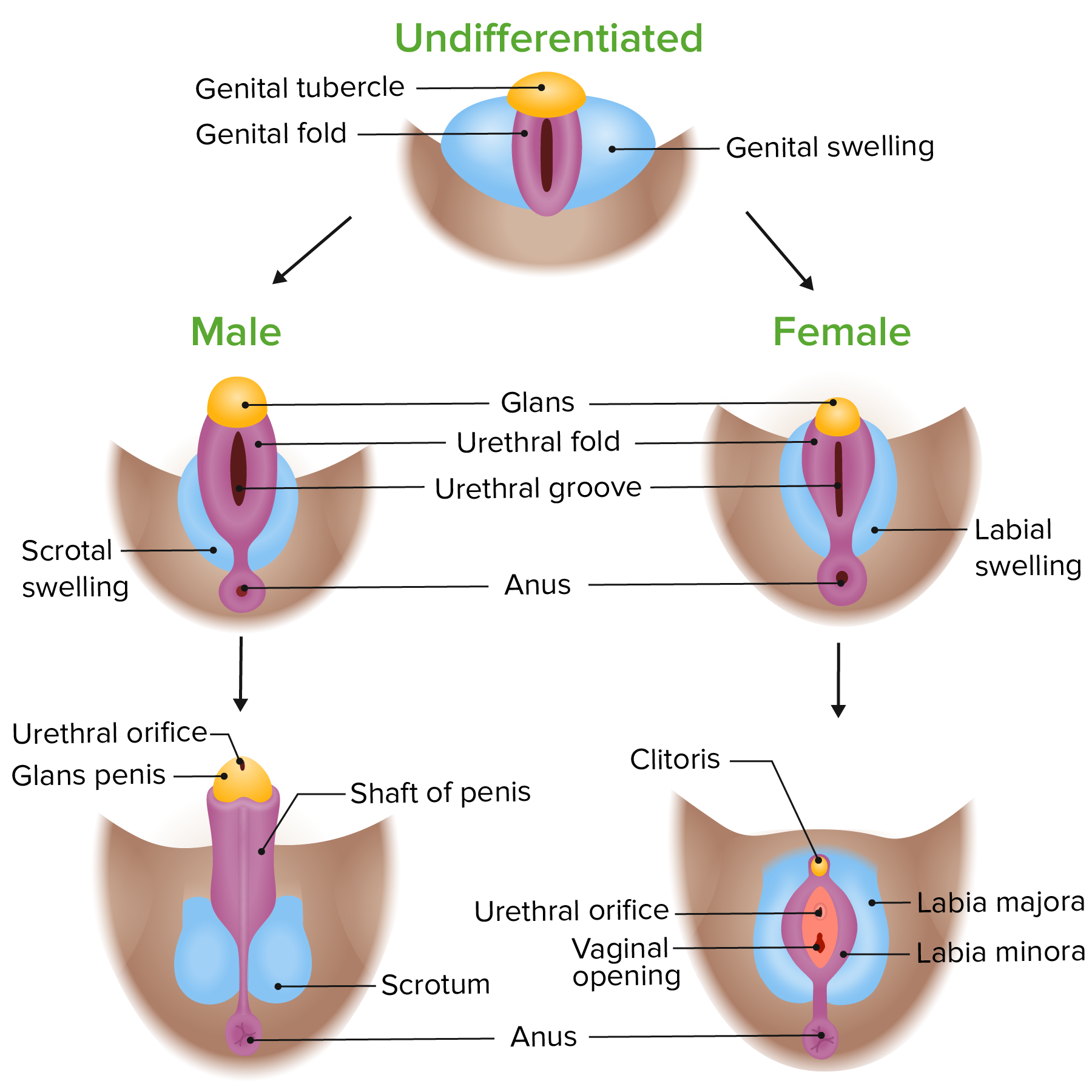
External genitalia develop from the undifferentiated genital tubercleGenital TubercleDevelopment of the Urogenital System, genital swellingSwellingInflammation, and genital foldsGenital foldsDevelopment of the Urogenital System based on the presence or absence of testosteroneTestosteroneA potent androgenic steroid and major product secreted by the leydig cells of the testis. Its production is stimulated by luteinizing hormone from the pituitary gland. In turn, testosterone exerts feedback control of the pituitary LH and FSH secretion. Depending on the tissues, testosterone can be further converted to dihydrotestosterone or estradiol.Androgens and Antiandrogens.
Male: testosteroneTestosteroneA potent androgenic steroid and major product secreted by the leydig cells of the testis. Its production is stimulated by luteinizing hormone from the pituitary gland. In turn, testosterone exerts feedback control of the pituitary LH and FSH secretion. Depending on the tissues, testosterone can be further converted to dihydrotestosterone or estradiol.Androgens and Antiandrogens → penisPenisThe penis is the male organ of copulation and micturition. The organ is composed of a root, body, and glans. The root is attached to the pubic bone by the crura penis. The body consists of the 2 parallel corpora cavernosa and the corpus spongiosum. The glans is ensheathed by the prepuce or foreskin. Penis: Anatomy and scrotumScrotumA cutaneous pouch of skin containing the testicles and spermatic cords.Testicles: Anatomy
Female: lack of testosteroneTestosteroneA potent androgenic steroid and major product secreted by the leydig cells of the testis. Its production is stimulated by luteinizing hormone from the pituitary gland. In turn, testosterone exerts feedback control of the pituitary LH and FSH secretion. Depending on the tissues, testosterone can be further converted to dihydrotestosterone or estradiol.Androgens and Antiandrogens → clitorisClitorisAn erectile structure homologous with the penis, situated beneath the anterior labial commissure, partially hidden between the anterior ends of the labia minora.Vagina, Vulva, and Pelvic Floor: Anatomy, labia majoraLabia majoraVagina, Vulva, and Pelvic Floor: Anatomy, labia minoraLabia minoraVagina, Vulva, and Pelvic Floor: Anatomy
Secondary sexual characteristicsSecondary Sexual CharacteristicsPrecocious Pubertydevelop based on the hormonal milieu at pubertyPubertyPuberty is a complex series of physical, psychosocial, and cognitive transitions usually experienced by adolescents (11-19 years of age). Puberty is marked by a growth in stature and the development of secondary sexual characteristics, achievement of fertility, and changes in most body systems.Puberty.
Androgenic characteristics: due to the presence of testosteroneTestosteroneA potent androgenic steroid and major product secreted by the leydig cells of the testis. Its production is stimulated by luteinizing hormone from the pituitary gland. In turn, testosterone exerts feedback control of the pituitary LH and FSH secretion. Depending on the tissues, testosterone can be further converted to dihydrotestosterone or estradiol.Androgens and Antiandrogens and/or dihydrotestosteroneDihydrotestosteroneA potent androgenic metabolite of testosterone. It is produced by the action of the enzyme 3-oxo-5-alpha-steroid 4-dehydrogenase.Gonadal Hormones (DHTDHTA potent androgenic metabolite of testosterone. It is produced by the action of the enzyme 3-oxo-5-alpha-steroid 4-dehydrogenase.Gonadal Hormones)
Pubic and axillary hair
Facial and body hair in an androgenic distribution and qualityQualityActivities and programs intended to assure or improve the quality of care in either a defined medical setting or a program. The concept includes the assessment or evaluation of the quality of care; identification of problems or shortcomings in the delivery of care; designing activities to overcome these deficiencies; and follow-up monitoring to ensure effectiveness of corrective steps.Quality Measurement and Improvement (dark and coarse)
Deepening of the voice
↑ Muscle massMassThree-dimensional lesion that occupies a space within the breastImaging of the Breast
Estrogenic characteristics result from the presence of estrogenEstrogenCompounds that interact with estrogen receptors in target tissues to bring about the effects similar to those of estradiol. Estrogens stimulate the female reproductive organs, and the development of secondary female sex characteristics. Estrogenic chemicals include natural, synthetic, steroidal, or non-steroidal compounds.Ovaries: Anatomy:
Breast development
Wider hips
Sex differentiation
Image by Lecturio.
Phenotypic differentiation of the external genitalia in male and female embryos
Image by Lecturio.
Typical male development
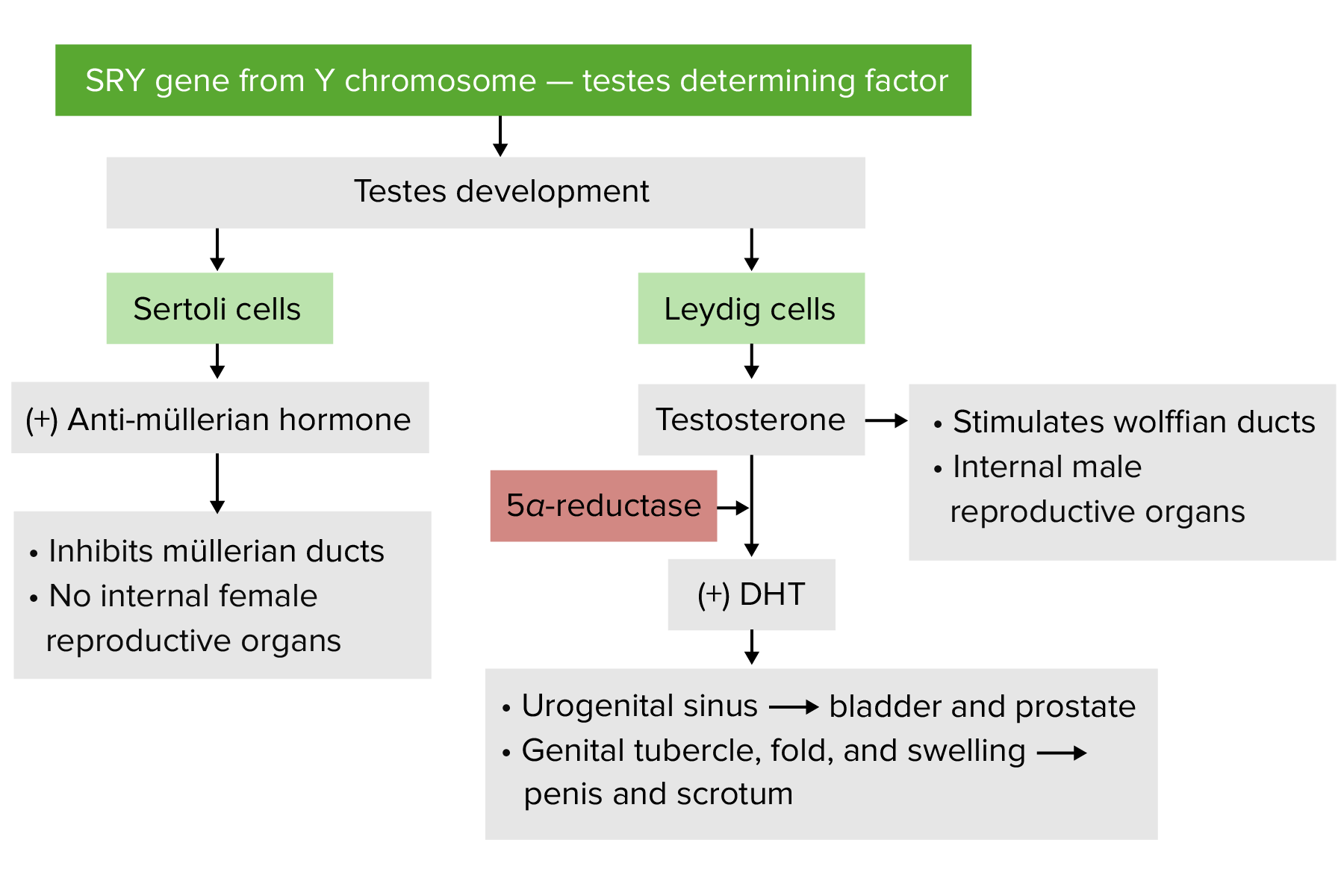
Male development begins due to the presence of the sex-determining region of the Y chromosomeY chromosomeThe male sex chromosome, being the differential sex chromosome carried by half the male gametes and none of the female gametes in humans and in some other male-heterogametic species in which the homologue of the X chromosome has been retained.Basic Terms of GeneticsgeneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics (SRYgeneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics).
SRYgeneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics→ produces SRY proteinSRY proteinDevelopment of the Urogenital System(also known as testis-determining factor):
Seminiferous tubulesSeminiferous TubulesThe convoluted tubules in the testis where sperm are produced (spermatogenesis) and conveyed to the rete testis. Spermatogenic tubules are composed of developing germ cells and the supporting sertoli cells.Testicles: Anatomy
Leydig cellsLeydig CellsSteroid-producing cells in the interstitial tissue of the testis. They are under the regulation of pituitary hormones; luteinizing hormone; or interstitial cell-stimulating hormone. Testosterone is the major androgen (androgens) produced.Testicles: Anatomy
Sertoli cellsSertoli CellsSupporting cells projecting inward from the basement membrane of seminiferous tubules. They surround and nourish the developing male germ cells and secrete androgen-binding protein and hormones such as anti-mullerian hormone. The tight junctions of sertoli cells with the spermatogonia and spermatocytes provide a blood-testis barrier.Testicles: Anatomy
Suppresses the differentiation of ovariesOvariesOvaries are the paired gonads of the female reproductive system that contain haploid gametes known as oocytes. The ovaries are located intraperitoneally in the pelvis, just posterior to the broad ligament, and are connected to the pelvic sidewall and to the uterus by ligaments. These organs function to secrete hormones (estrogen and progesterone) and to produce the female germ cells (oocytes).Ovaries: Anatomy
AMHAMHA glycoprotein that causes regression of mullerian ducts. It is produced by sertoli cells of the testes. In the absence of this hormone, the mullerian ducts develop into structures of the female reproductive tract. In males, defects of this hormone result in persistent mullerian duct, a form of male pseudohermaphroditism.Primary Amenorrhea
TestosteroneTestosteroneA potent androgenic steroid and major product secreted by the leydig cells of the testis. Its production is stimulated by luteinizing hormone from the pituitary gland. In turn, testosterone exerts feedback control of the pituitary LH and FSH secretion. Depending on the tissues, testosterone can be further converted to dihydrotestosterone or estradiol.Androgens and Antiandrogens
DHTDHTA potent androgenic metabolite of testosterone. It is produced by the action of the enzyme 3-oxo-5-alpha-steroid 4-dehydrogenase.Gonadal Hormones
The above hormonesHormonesHormones are messenger molecules that are synthesized in one part of the body and move through the bloodstream to exert specific regulatory effects on another part of the body. Hormones play critical roles in coordinating cellular activities throughout the body in response to the constant changes in both the internal and external environments. Hormones: Overview and Types cause:
Stimulation of the Wolffian ductsWolffian ductsA pair of excretory ducts of the middle kidneys of an embryo, also called mesonephric ducts. In higher vertebrates, wolffian ducts persist in the male forming vas deferens, but atrophy into vestigial structures in the female.Primary Amenorrhea (around the 8th week) to differentiate into the epididymisEpididymisThe convoluted cordlike structure attached to the posterior of the testis. Epididymis consists of the head (caput), the body (corpus), and the tail (cauda). A network of ducts leaving the testis joins into a common epididymal tubule proper which provides the transport, storage, and maturation of spermatozoa.Testicles: Anatomy, vas deferensVas DeferensThe excretory duct of the testes that carries spermatozoa. It rises from the scrotum and joins the seminal vesicles to form the ejaculatory duct.Testicles: Anatomy, seminal vesiclesVesiclesFemale Genitourinary Examination, and ejaculatory ductsEjaculatory DuctsPaired ducts in the human male through which semen is ejaculated into the urethra.
Differentiation of the prostateProstateThe prostate is a gland in the male reproductive system. The gland surrounds the bladder neck and a portion of the urethra. The prostate is an exocrine gland that produces a weakly acidic secretion, which accounts for roughly 20% of the seminal fluid. and external genitalia
The process of differentiation is largely completed by 12 weeks of gestation.
Male development
DHT: dihydrotestosterone Image by Lecturio.
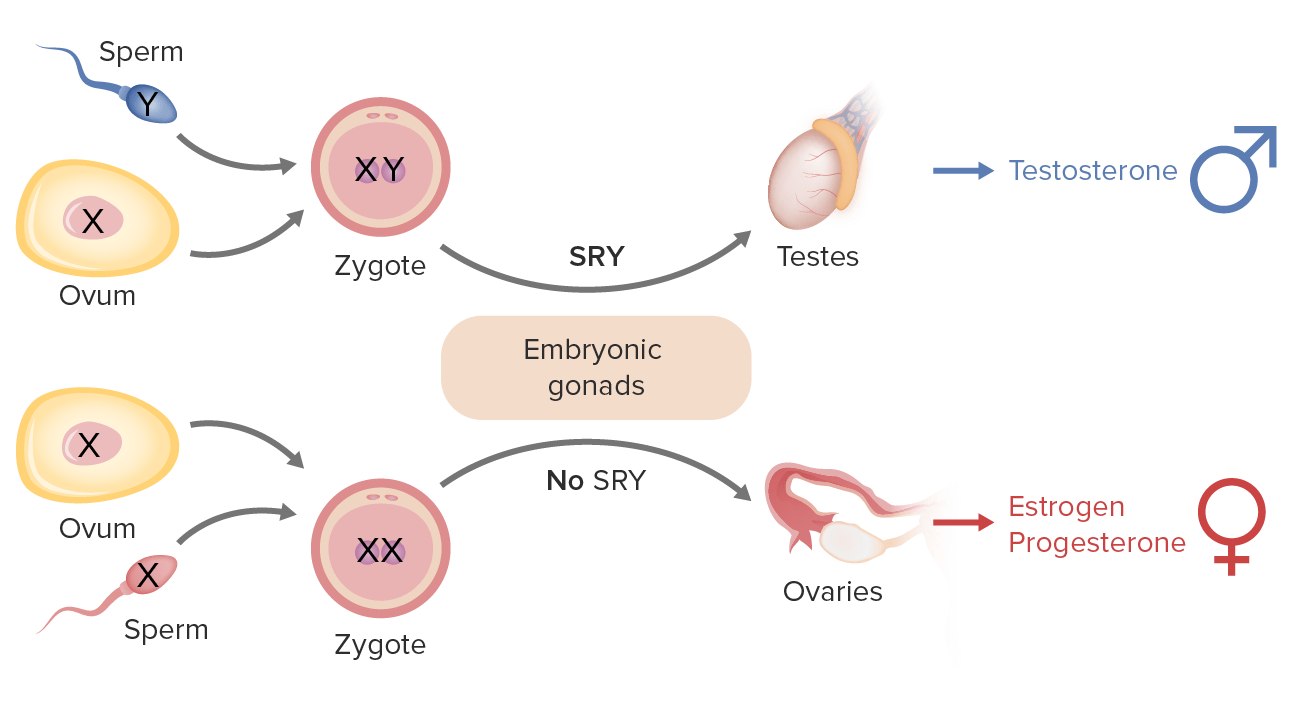
Sex determination in humans: The Y chromosome includes the SRY gene (Sex-determining Region Y) that codes for testis-determining factor (TDF), causing embryonic gonads to form into testes (male gonads) and lead to male development. In the absence of the TDF protein (i.e., no Y chromosome), the embryonic gonads develop into ovaries (female gonads). Females possess 2 copies of the X chromosome (XX); males possess 1 X and 1 Y (shorter) chromosome (XY).
Image by Lecturio.
Typical female development
Development of the ovary from the bipotent gonad requires:
Presence of several genesGenesA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.DNA Types and Structure found on chromosomeChromosomeIn a prokaryotic cell or in the nucleus of a eukaryotic cell, a structure consisting of or containing DNA which carries the genetic information essential to the cell.Basic Terms of Genetics 1, including:
WNT4
R spondin 1 (Rspo1)
Begins around 10 weeks of development
Differentiation and development of the Müllerian ducts and external genitalia occur when testicular androgensAndrogensAndrogens are naturally occurring steroid hormones responsible for development and maintenance of the male sex characteristics, including penile, scrotal, and clitoral growth, development of sexual hair, deepening of the voice, and musculoskeletal growth. Androgens and Antiandrogens and AMHAMHA glycoprotein that causes regression of mullerian ducts. It is produced by sertoli cells of the testes. In the absence of this hormone, the mullerian ducts develop into structures of the female reproductive tract. In males, defects of this hormone result in persistent mullerian duct, a form of male pseudohermaphroditism.Primary Amenorrhea are absent.
Embryos develop in the “female hormonal environment” within their mother; thus, it is unclear whether hormonesHormonesHormones are messenger molecules that are synthesized in one part of the body and move through the bloodstream to exert specific regulatory effects on another part of the body. Hormones play critical roles in coordinating cellular activities throughout the body in response to the constant changes in both the internal and external environments. Hormones: Overview and Types produced by the fetus are required for their development.
Androgen insensitivity syndromeAndrogen insensitivity syndromeAndrogen insensitivity syndrome (AIS) is an X-linked recessive condition in which a genetic mutation affects the function of androgen receptors, resulting in complete (CAIS), partial (PAIS), or mild (MAIS) resistance to testosterone. All individuals with AIS have a 46,XY karyotype; however, phenotypes vary and include phenotypic female, virilized female, undervirilized male, and phenotypic male individuals. Androgen Insensitivity Syndrome (AISAISScoliosis)
Decreased or no response to androgensAndrogensAndrogens are naturally occurring steroid hormones responsible for development and maintenance of the male sex characteristics, including penile, scrotal, and clitoral growth, development of sexual hair, deepening of the voice, and musculoskeletal growth. Androgens and Antiandrogens
Due to mutations affecting the quantity or function of androgen receptorsReceptorsReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors
↑↑ Serum testosteroneTestosteroneA potent androgenic steroid and major product secreted by the leydig cells of the testis. Its production is stimulated by luteinizing hormone from the pituitary gland. In turn, testosterone exerts feedback control of the pituitary LH and FSH secretion. Depending on the tissues, testosterone can be further converted to dihydrotestosterone or estradiol.Androgens and Antiandrogens levels
Typically present in adolescence as primary amenorrheaAmenorrheaAbsence of menstruation.Congenital Malformations of the Female Reproductive System, well-developed breastsBreastsThe breasts are found on the anterior thoracic wall and consist of mammary glands surrounded by connective tissue. The mammary glands are modified apocrine sweat glands that produce milk, which serves as nutrition for infants. Breasts are rudimentary and usually nonfunctioning in men. Breasts: Anatomy, and sparse pubic hair
5-α-reductase deficiency
Loss-of-function mutationMutationGenetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations in 5-α-reductase geneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics impairs conversion of testosteroneTestosteroneA potent androgenic steroid and major product secreted by the leydig cells of the testis. Its production is stimulated by luteinizing hormone from the pituitary gland. In turn, testosterone exerts feedback control of the pituitary LH and FSH secretion. Depending on the tissues, testosterone can be further converted to dihydrotestosterone or estradiol.Androgens and Antiandrogens to dihydrotestosteroneDihydrotestosteroneA potent androgenic metabolite of testosterone. It is produced by the action of the enzyme 3-oxo-5-alpha-steroid 4-dehydrogenase.Gonadal Hormones (DHTDHTA potent androgenic metabolite of testosterone. It is produced by the action of the enzyme 3-oxo-5-alpha-steroid 4-dehydrogenase.Gonadal Hormones).
Normal or ↑ testosteroneTestosteroneA potent androgenic steroid and major product secreted by the leydig cells of the testis. Its production is stimulated by luteinizing hormone from the pituitary gland. In turn, testosterone exerts feedback control of the pituitary LH and FSH secretion. Depending on the tissues, testosterone can be further converted to dihydrotestosterone or estradiol.Androgens and Antiandrogens with ↓ DHTDHTA potent androgenic metabolite of testosterone. It is produced by the action of the enzyme 3-oxo-5-alpha-steroid 4-dehydrogenase.Gonadal Hormones
AromataseAromataseAn enzyme that catalyzes the desaturation (aromatization) of the ring a of C19 androgens and converts them to C18 estrogens. In this process, the 19-methyl is removed. This enzyme is membrane-bound, located in the endoplasmic reticulum of estrogen-producing cells of ovaries, placenta, testes, adipose, and brain tissues. Aromatase is encoded by the cyp19 gene, and functions in complex with NADPH-ferrihemoprotein reductase in the cytochrome p450 system.Adipose Tissue: Histology deficiency
↓ AromataseAromataseAn enzyme that catalyzes the desaturation (aromatization) of the ring a of C19 androgens and converts them to C18 estrogens. In this process, the 19-methyl is removed. This enzyme is membrane-bound, located in the endoplasmic reticulum of estrogen-producing cells of ovaries, placenta, testes, adipose, and brain tissues. Aromatase is encoded by the cyp19 gene, and functions in complex with NADPH-ferrihemoprotein reductase in the cytochrome p450 system.Adipose Tissue: Histology → ↓ conversion of testosteroneTestosteroneA potent androgenic steroid and major product secreted by the leydig cells of the testis. Its production is stimulated by luteinizing hormone from the pituitary gland. In turn, testosterone exerts feedback control of the pituitary LH and FSH secretion. Depending on the tissues, testosterone can be further converted to dihydrotestosterone or estradiol.Androgens and Antiandrogens into estrogenEstrogenCompounds that interact with estrogen receptors in target tissues to bring about the effects similar to those of estradiol. Estrogens stimulate the female reproductive organs, and the development of secondary female sex characteristics. Estrogenic chemicals include natural, synthetic, steroidal, or non-steroidal compounds.Ovaries: Anatomy
Individuals will have:
↓ EstrogenEstrogenCompounds that interact with estrogen receptors in target tissues to bring about the effects similar to those of estradiol. Estrogens stimulate the female reproductive organs, and the development of secondary female sex characteristics. Estrogenic chemicals include natural, synthetic, steroidal, or non-steroidal compounds.Ovaries: Anatomy
↑ TestosteroneTestosteroneA potent androgenic steroid and major product secreted by the leydig cells of the testis. Its production is stimulated by luteinizing hormone from the pituitary gland. In turn, testosterone exerts feedback control of the pituitary LH and FSH secretion. Depending on the tissues, testosterone can be further converted to dihydrotestosterone or estradiol.Androgens and Antiandrogens
All are prone to effects of ↑ testosteroneTestosteroneA potent androgenic steroid and major product secreted by the leydig cells of the testis. Its production is stimulated by luteinizing hormone from the pituitary gland. In turn, testosterone exerts feedback control of the pituitary LH and FSH secretion. Depending on the tissues, testosterone can be further converted to dihydrotestosterone or estradiol.Androgens and Antiandrogens (e.g., fertility issues, dyslipidemia, glucoseGlucoseA primary source of energy for living organisms. It is naturally occurring and is found in fruits and other parts of plants in its free state. It is used therapeutically in fluid and nutrient replacement.Lactose Intolerance intolerance).
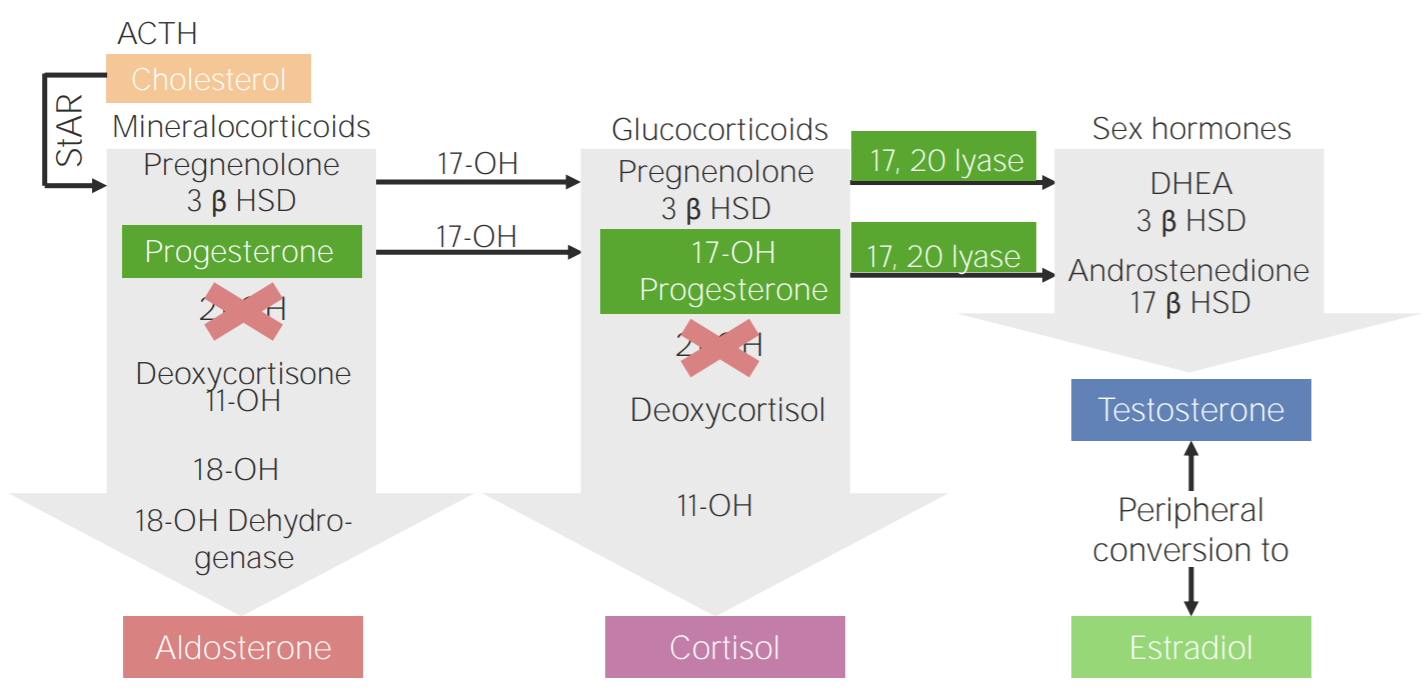
Congenital adrenal hyperplasiaHyperplasiaAn increase in the number of cells in a tissue or organ without tumor formation. It differs from hypertrophy, which is an increase in bulk without an increase in the number of cells.Cellular Adaptation
Due to deficiencies in 1 of the enzymesEnzymesEnzymes are complex protein biocatalysts that accelerate chemical reactions without being consumed by them. Due to the body’s constant metabolic needs, the absence of enzymes would make life unsustainable, as reactions would occur too slowly without these molecules. Basics of Enzymes involved in the steroidogenesis pathway
Intermediate substrates are shunted toward testosteroneTestosteroneA potent androgenic steroid and major product secreted by the leydig cells of the testis. Its production is stimulated by luteinizing hormone from the pituitary gland. In turn, testosterone exerts feedback control of the pituitary LH and FSH secretion. Depending on the tissues, testosterone can be further converted to dihydrotestosterone or estradiol.Androgens and Antiandrogens production (see diagram below).
Leads to ↓ cortisolCortisolGlucocorticoids, ↓ aldosteroneAldosteroneA hormone secreted by the adrenal cortex that regulates electrolyte and water balance by increasing the renal retention of sodium and the excretion of potassium.Hyperkalemia, ↑ ACTH, and ↑ testosteroneTestosteroneA potent androgenic steroid and major product secreted by the leydig cells of the testis. Its production is stimulated by luteinizing hormone from the pituitary gland. In turn, testosterone exerts feedback control of the pituitary LH and FSH secretion. Depending on the tissues, testosterone can be further converted to dihydrotestosterone or estradiol.Androgens and Antiandrogens
Normal Wolffian structures and phenotypic male genitalia
Salt-wasting crisis (1-2 weeks of life)
Early virilization
All are prone to effects of ↑ testosteroneTestosteroneA potent androgenic steroid and major product secreted by the leydig cells of the testis. Its production is stimulated by luteinizing hormone from the pituitary gland. In turn, testosterone exerts feedback control of the pituitary LH and FSH secretion. Depending on the tissues, testosterone can be further converted to dihydrotestosterone or estradiol.Androgens and Antiandrogens and adrenal tumors.
Kallmann syndromeKallmann syndromeKallmann syndrome (KS), also called olfacto-genital syndrome, is a genetic condition that causes hypogonadotropic hypogonadism due to decreased secretion of gonadotropin-releasing hormone (GnRH) by the hypothalamus. The lack of sex hormones results in impaired pubertal development. Kallmann Syndrome
Decreased secretionSecretionCoagulation Studies or lack of gonadotropin-releasing hormoneGonadotropin-releasing hormoneA decapeptide that stimulates the synthesis and secretion of both pituitary gonadotropins, luteinizing hormone and follicle stimulating hormone. Gnrh is produced by neurons in the septum preoptic area of the hypothalamus and released into the pituitary portal blood, leading to stimulation of gonadotrophs in the anterior pituitary gland.Puberty (GnRH)
Leads to a lack of allsexSexThe totality of characteristics of reproductive structure, functions, phenotype, and genotype, differentiating the male from the female organism.Gender DysphoriahormonesHormonesHormones are messenger molecules that are synthesized in one part of the body and move through the bloodstream to exert specific regulatory effects on another part of the body. Hormones play critical roles in coordinating cellular activities throughout the body in response to the constant changes in both the internal and external environments. Hormones: Overview and Types
Affects both sexes
Can be due to mutations in a variety of genesGenesA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.DNA Types and Structure
Presents with absence of sexual development at pubertyPubertyPuberty is a complex series of physical, psychosocial, and cognitive transitions usually experienced by adolescents (11-19 years of age). Puberty is marked by a growth in stature and the development of secondary sexual characteristics, achievement of fertility, and changes in most body systems.Puberty due to lack of sexSexThe totality of characteristics of reproductive structure, functions, phenotype, and genotype, differentiating the male from the female organism.Gender DysphoriahormonesHormonesHormones are messenger molecules that are synthesized in one part of the body and move through the bloodstream to exert specific regulatory effects on another part of the body. Hormones play critical roles in coordinating cellular activities throughout the body in response to the constant changes in both the internal and external environments. Hormones: Overview and Types
Key finding: associated with anosmiaAnosmiaComplete or severe loss of the subjective sense of smell. Loss of smell may be caused by many factors such as a cold, allergy, olfactory nerve diseases, viral respiratory tract infections (e.g., COVID-19), aging and various neurological disorders (e.g., Alzheimer disease).Cranial Nerve Palsies (absent sense of smellSmellThe sense of smell, or olfaction, begins in a small area on the roof of the nasal cavity, which is covered in specialized mucosa. From there, the olfactory nerve transmits the sensory perception of smell via the olfactory pathway. This pathway is composed of the olfactory cells and bulb, the tractus and striae olfactoriae, and the primary olfactory cortex and amygdala.Olfaction: Anatomy)
Other anomalies may be present, including cleft lipCleft lipThe embryological development of craniofacial structures is an intricate sequential process involving tissue growth and directed cell apoptosis. Disruption of any step in this process may result in the formation of a cleft lip alone or in combination with a cleft palate. As the most common craniofacial malformation of the newborn, the diagnosis of a cleft is clinical and usually apparent at birth. Cleft Lip and Cleft Palate/palatePalateThe palate is the structure that forms the roof of the mouth and floor of the nasal cavity. This structure is divided into soft and hard palates. Palate: Anatomy, hearing lossHearing lossHearing loss, also known as hearing impairment, is any degree of impairment in the ability to apprehend sound as determined by audiometry to be below normal hearing thresholds. Clinical presentation may occur at birth or as a gradual loss of hearing with age, including a short-term or sudden loss at any point. Hearing Loss, syndactylySyndactylyA congenital anomaly of the hand or foot, marked by the webbing between adjacent fingers or toes. Syndactylies are classified as complete or incomplete by the degree of joining. Syndactylies can also be simple or complex. Simple syndactyly indicates joining of only skin or soft tissue; complex syndactyly marks joining of bony elements.Development of the Limbs, and renal agenesisAgenesisTeratogenic Birth Defects.
46,XX gonadal dysgenesisGonadal dysgenesisA number of syndromes with defective gonadal developments such as streak gonads and dysgenetic testes or ovaries. The spectrum of gonadal and sexual abnormalities is reflected in their varied sex chromosome (sex chromosomes) constitution as shown by the karyotypes of 45, X monosomy (Turner syndrome); 46, XX (gonadal dysgenesis, 46xx); 46, XY (gonadal dysgenesis, 46, xy); and sex chromosome mosaicism. Their phenotypes range from female, through ambiguous, to male. This concept includes gonadal agenesis.Wilms Tumor
Non-functional ovarian tissue → ↓↓ estrogenEstrogenCompounds that interact with estrogen receptors in target tissues to bring about the effects similar to those of estradiol. Estrogens stimulate the female reproductive organs, and the development of secondary female sex characteristics. Estrogenic chemicals include natural, synthetic, steroidal, or non-steroidal compounds.Ovaries: Anatomy
Due to various mutations affecting ovarian development
Swyer syndromeSwyer syndromeSwyer syndrome is a disorder of sex development caused by a defect in the SRY gene on chromosome Y. The syndrome is characterized by complete testicular dysgenesis in an individual who has a 46,XY karyotype and is phenotypically female. Swyer Syndrome
46,XY individuals with non-functional gonadal tissue → ↓ testosteroneTestosteroneA potent androgenic steroid and major product secreted by the leydig cells of the testis. Its production is stimulated by luteinizing hormone from the pituitary gland. In turn, testosterone exerts feedback control of the pituitary LH and FSH secretion. Depending on the tissues, testosterone can be further converted to dihydrotestosterone or estradiol.Androgens and Antiandrogens and ↓ estrogenEstrogenCompounds that interact with estrogen receptors in target tissues to bring about the effects similar to those of estradiol. Estrogens stimulate the female reproductive organs, and the development of secondary female sex characteristics. Estrogenic chemicals include natural, synthetic, steroidal, or non-steroidal compounds.Ovaries: Anatomy
AMHAMHA glycoprotein that causes regression of mullerian ducts. It is produced by sertoli cells of the testes. In the absence of this hormone, the mullerian ducts develop into structures of the female reproductive tract. In males, defects of this hormone result in persistent mullerian duct, a form of male pseudohermaphroditism.Primary Amenorrhea and testosteroneTestosteroneA potent androgenic steroid and major product secreted by the leydig cells of the testis. Its production is stimulated by luteinizing hormone from the pituitary gland. In turn, testosterone exerts feedback control of the pituitary LH and FSH secretion. Depending on the tissues, testosterone can be further converted to dihydrotestosterone or estradiol.Androgens and Antiandrogens are not secreted in developing embryoEmbryoThe entity of a developing mammal, generally from the cleavage of a zygote to the end of embryonic differentiation of basic structures. For the human embryo, this represents the first two months of intrauterine development preceding the stages of the fetus.Fertilization and First Week → development of uterusUterusThe uterus, cervix, and fallopian tubes are part of the internal female reproductive system. The uterus has a thick wall made of smooth muscle (the myometrium) and an inner mucosal layer (the endometrium). The most inferior portion of the uterus is the cervix, which connects the uterine cavity to the vagina.Uterus, Cervix, and Fallopian Tubes: Anatomy, vaginaVaginaThe vagina is the female genital canal, extending from the vulva externally to the cervix uteri internally. The structures have sexual, reproductive, and urinary functions and a rich blood supply, mainly arising from the internal iliac artery.Vagina, Vulva, and Pelvic Floor: Anatomy, and female genitalia
Mutations may be on an X, Y (SRY region), or autosomal chromosomeChromosomeIn a prokaryotic cell or in the nucleus of a eukaryotic cell, a structure consisting of or containing DNA which carries the genetic information essential to the cell.Basic Terms of Genetics.
True hermaphroditismTrue hermaphroditismTrue hermaphroditism, or ovotesticular disorder of sexual development (ODSD), is characterized by the presence of an ovotesticular gonad that contains both ovarian and testicular elements. Individuals are usually born with ambiguous genitalia, but the diagnosis is rarely confirmed before puberty. The most common karyotype is 46,XX, and less often, 46,XY can be identified.Ovotesticular Disorder of Sexual Development (ODSD)
PatientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship have at least 1 ovotestis gonad that contains both ovarian and testicular elements.
May have a normal ovary or testis on the contralateral side
KaryotypeKaryotypeThe full set of chromosomes presented as a systematized array of metaphase chromosomes from a photomicrograph of a single cell nucleus arranged in pairs in descending order of size and according to the position of the centromere.Congenital Malformations of the Female Reproductive System may be either 46,XX (approximately 80%), 46,XY, or 46,XX/XY mosaic (rare).
Fallopian tubesFallopian tubesThe uterus, cervix, and fallopian tubes are part of the internal female reproductive system. The fallopian tubes receive an ovum after ovulation and help move it and/or a fertilized embryo toward the uterus via ciliated cells lining the tubes and peristaltic movements of its smooth muscle. Uterus, Cervix, and Fallopian Tubes: Anatomy may develop beside an ovary or ovotestis (60%‒70% of the time).
Vas deferensVas DeferensThe excretory duct of the testes that carries spermatozoa. It rises from the scrotum and joins the seminal vesicles to form the ejaculatory duct.Testicles: Anatomy and epididymisEpididymisThe convoluted cordlike structure attached to the posterior of the testis. Epididymis consists of the head (caput), the body (corpus), and the tail (cauda). A network of ducts leaving the testis joins into a common epididymal tubule proper which provides the transport, storage, and maturation of spermatozoa.Testicles: Anatomy develop beside a testicle.
UterusUterusThe uterus, cervix, and fallopian tubes are part of the internal female reproductive system. The uterus has a thick wall made of smooth muscle (the myometrium) and an inner mucosal layer (the endometrium). The most inferior portion of the uterus is the cervix, which connects the uterine cavity to the vagina.Uterus, Cervix, and Fallopian Tubes: Anatomy may develop if an ovary is present.
Turner syndromeTurner syndromeTurner syndrome is a genetic condition affecting women, in which 1 X chromosome is partly or completely missing. The classic result is the karyotype 45,XO with a female phenotype. Turner syndrome is associated with decreased sex hormone levels and is the most common cause of primary amenorrhea.Turner Syndrome
Chromosomal anomaly with a partially or completely missing X chromosomeX chromosomeThe female sex chromosome, being the differential sex chromosome carried by half the male gametes and all female gametes in human and other male-heterogametic species.Basic Terms of Genetics
Occurs due to nondisjunctionNondisjunctionThe failure of homologous chromosomes or chromatids to segregate during mitosis or meiosis with the result that one daughter cell has both of a pair of parental chromosomes or chromatids and the other has none.Basic Terms of Genetics during meiosisMeiosisThe creation of eukaryotic gametes involves a DNA replication phase followed by 2 cellular division stages: meiosis I and meiosis II. Meiosis I separates homologous chromosomes into separate cells (1n, 2c), while meiosis II separates sister chromatids into gametes (1n, 1c). Meiosis or mitosisMitosisA type of cell nucleus division by means of which the two daughter nuclei normally receive identical complements of the number of chromosomes of the somatic cells of the species.Cell Cycle
Affected individuals are 45,X0.
Phenotypic female genitalia
Müllerian structures present
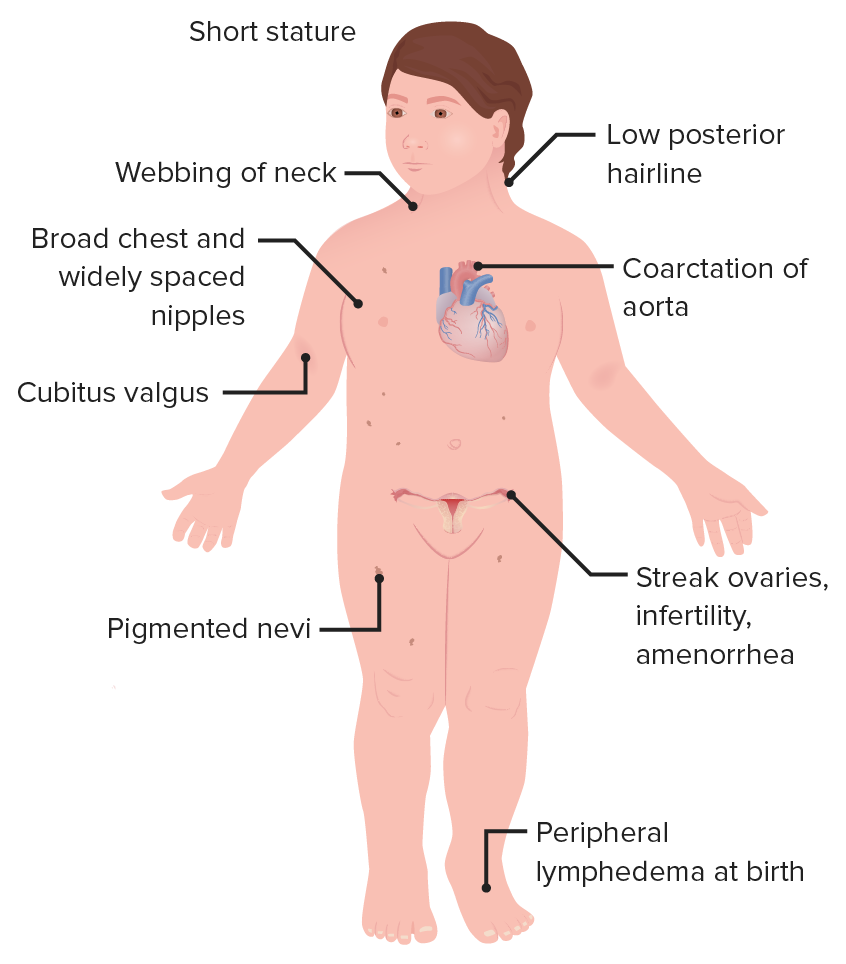
Characteristic appearance including short stature, webbed neckWebbed neckTurner Syndrome, shield chest, and low hairline
Multiple medical anomalies involving the cardiac, renal, reproductive, skeletal, and lymphatic systems
Klinefelter syndromeKlinefelter syndromeKlinefelter syndrome is a chromosomal aneuploidy characterized by the presence of 1 or more extra X chromosomes in a male karyotype, most commonly leading to karyotype 47,XXY. Klinefelter syndrome is associated with decreased levels of testosterone and is the most common cause of congenital hypogonadism. Klinefelter Syndrome
Chromosomal anomaly characterized by 2 or more X chromosomesChromosomesIn a prokaryotic cell or in the nucleus of a eukaryotic cell, a structure consisting of or containing DNA which carries the genetic information essential to the cell.DNA Types and Structure in a male karyotypeKaryotypeThe full set of chromosomes presented as a systematized array of metaphase chromosomes from a photomicrograph of a single cell nucleus arranged in pairs in descending order of size and according to the position of the centromere.Congenital Malformations of the Female Reproductive System
Most commonly 47,XXYXXYKlinefelter syndrome is a chromosomal aneuploidy characterized by the presence of 1 or more extra X chromosomes in a male karyotype, most commonly leading to karyotype 47,XXY. Klinefelter syndrome is associated with decreased levels of testosterone and is the most common cause of congenital hypogonadism.Klinefelter Syndrome
Other possible karyotypes: 48,XXXY and 48,XXYY
↓ TestosteroneTestosteroneA potent androgenic steroid and major product secreted by the leydig cells of the testis. Its production is stimulated by luteinizing hormone from the pituitary gland. In turn, testosterone exerts feedback control of the pituitary LH and FSH secretion. Depending on the tissues, testosterone can be further converted to dihydrotestosterone or estradiol.Androgens and Antiandrogens and ↑ estrogenEstrogenCompounds that interact with estrogen receptors in target tissues to bring about the effects similar to those of estradiol. Estrogens stimulate the female reproductive organs, and the development of secondary female sex characteristics. Estrogenic chemicals include natural, synthetic, steroidal, or non-steroidal compounds.Ovaries: Anatomy compared to typical levels in men
Presents in adolescence with small testesTestesGonadal Hormones, ↓ body hair, gynecomastiaGynecomastiaGynecomastia is a benign proliferation of male breast glandular ductal tissue, usually bilateral, caused by increased estrogen activity, decreased testosterone activity, or medications. The condition is common and physiological in neonates, adolescent boys, and elderly men. Gynecomastia, and infertilityInfertilityInfertility is the inability to conceive in the context of regular intercourse. The most common causes of infertility in women are related to ovulatory dysfunction or tubal obstruction, whereas, in men, abnormal sperm is a common cause. Infertility
Most common cause of hypogonadismHypogonadismHypogonadism is a condition characterized by reduced or no sex hormone production by the testes or ovaries. Hypogonadism can result from primary (hypergonadotropic) or secondary (hypogonadotropic) failure. Symptoms include infertility, increased risk of osteoporosis, erectile dysfunction, decreased libido, and regression (or absence) of secondary sexual characteristics.Hypogonadism in men
21-hydroxylase deficiency: Diagram of the pathophysiology of congenital adrenal hyperplasia due to 21-hydroxylase deficiency
The differential diagnosis for disorders of sexual development typically includes many of the conditions listed above. The general diagnostic approach to help determine the appropriate diagnosis is listed below.
Exam
Genital examination:
Ambiguous genitaliaAmbiguous GenitaliaPrimary Amenorrhea is likely due to ↑ testosteroneTestosteroneA potent androgenic steroid and major product secreted by the leydig cells of the testis. Its production is stimulated by luteinizing hormone from the pituitary gland. In turn, testosterone exerts feedback control of the pituitary LH and FSH secretion. Depending on the tissues, testosterone can be further converted to dihydrotestosterone or estradiol.Androgens and Antiandrogens/DHTDHTA potent androgenic metabolite of testosterone. It is produced by the action of the enzyme 3-oxo-5-alpha-steroid 4-dehydrogenase.Gonadal Hormones in individuals without SRY (e.g., 46,XX) or ↓ testosteroneTestosteroneA potent androgenic steroid and major product secreted by the leydig cells of the testis. Its production is stimulated by luteinizing hormone from the pituitary gland. In turn, testosterone exerts feedback control of the pituitary LH and FSH secretion. Depending on the tissues, testosterone can be further converted to dihydrotestosterone or estradiol.Androgens and Antiandrogens in individuals with SRY (e.g., 46,XY)
Internal pelvic examination on phenotypic females:
Is a cervixCervixThe uterus, cervix, and fallopian tubes are part of the internal female reproductive system. The most inferior portion of the uterus is the cervix, which connects the uterine cavity to the vagina. Externally, the cervix is lined by stratified squamous cells; however, the cervical canal is lined by columnar epithelium.Uterus, Cervix, and Fallopian Tubes: Anatomy visible?
Is the uterusUterusThe uterus, cervix, and fallopian tubes are part of the internal female reproductive system. The uterus has a thick wall made of smooth muscle (the myometrium) and an inner mucosal layer (the endometrium). The most inferior portion of the uterus is the cervix, which connects the uterine cavity to the vagina.Uterus, Cervix, and Fallopian Tubes: Anatomy palpable?
Breast development = estrogenEstrogenCompounds that interact with estrogen receptors in target tissues to bring about the effects similar to those of estradiol. Estrogens stimulate the female reproductive organs, and the development of secondary female sex characteristics. Estrogenic chemicals include natural, synthetic, steroidal, or non-steroidal compounds.Ovaries: Anatomy present
Pubic and axillary hair = testosteroneTestosteroneA potent androgenic steroid and major product secreted by the leydig cells of the testis. Its production is stimulated by luteinizing hormone from the pituitary gland. In turn, testosterone exerts feedback control of the pituitary LH and FSH secretion. Depending on the tissues, testosterone can be further converted to dihydrotestosterone or estradiol.Androgens and Antiandrogens present
Facial/body hair, deepening of the voice = testosteroneTestosteroneA potent androgenic steroid and major product secreted by the leydig cells of the testis. Its production is stimulated by luteinizing hormone from the pituitary gland. In turn, testosterone exerts feedback control of the pituitary LH and FSH secretion. Depending on the tissues, testosterone can be further converted to dihydrotestosterone or estradiol.Androgens and Antiandrogens present
Look for findings associated with specific syndromes (e.g., webbed neckWebbed neckTurner Syndrome in Turner syndromeTurner syndromeTurner syndrome is a genetic condition affecting women, in which 1 X chromosome is partly or completely missing. The classic result is the karyotype 45,XO with a female phenotype. Turner syndrome is associated with decreased sex hormone levels and is the most common cause of primary amenorrhea.Turner Syndrome).
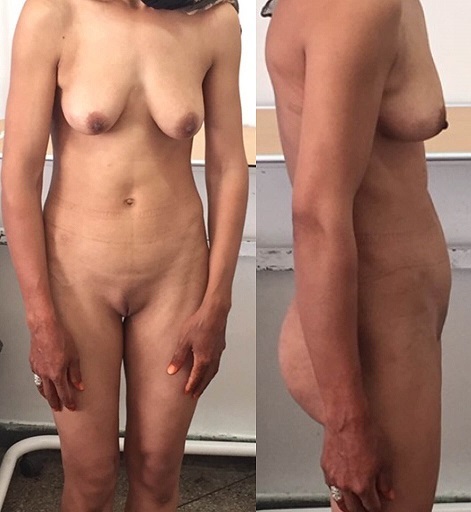
Androgen insensitivity syndrome: A 30-year-old patient with primary amenorrhea. On exam, well-developed breasts and absence of pubic hair are noted, indicating exposure to estrogen without significant testosterone exposure.
Image: “Complete androgen insensitivity syndrome or testicular feminization: Review of literature based on a case report” by The Pan African Medical Journal. License: CC BY 2.0
Characteristic features of a female with Turner syndrome
Image by Lecturio.
Laboratory
KaryotypeKaryotypeThe full set of chromosomes presented as a systematized array of metaphase chromosomes from a photomicrograph of a single cell nucleus arranged in pairs in descending order of size and according to the position of the centromere.Congenital Malformations of the Female Reproductive System with FISHFISHA type of in situ hybridization in which target sequences are stained with fluorescent dye so their location and size can be determined using fluorescence microscopy. This staining is sufficiently distinct that the hybridization signal can be seen both in metaphase spreads and in interphase nuclei.Chromosome Testing to assess the presence or absences of the SRYgeneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics
TestosteroneTestosteroneA potent androgenic steroid and major product secreted by the leydig cells of the testis. Its production is stimulated by luteinizing hormone from the pituitary gland. In turn, testosterone exerts feedback control of the pituitary LH and FSH secretion. Depending on the tissues, testosterone can be further converted to dihydrotestosterone or estradiol.Androgens and Antiandrogens:
Decreases in Kallman, Swyer, and Klinefelter syndromes
Increases or is normal in androgen insensitivity syndromeAndrogen insensitivity syndromeAndrogen insensitivity syndrome (AIS) is an X-linked recessive condition in which a genetic mutation affects the function of androgen receptors, resulting in complete (CAIS), partial (PAIS), or mild (MAIS) resistance to testosterone. All individuals with AIS have a 46,XY karyotype; however, phenotypes vary and include phenotypic female, virilized female, undervirilized male, and phenotypic male individuals. Androgen Insensitivity Syndrome (AISAISScoliosis), 5α-reductase deficiency, aromataseAromataseAn enzyme that catalyzes the desaturation (aromatization) of the ring a of C19 androgens and converts them to C18 estrogens. In this process, the 19-methyl is removed. This enzyme is membrane-bound, located in the endoplasmic reticulum of estrogen-producing cells of ovaries, placenta, testes, adipose, and brain tissues. Aromatase is encoded by the cyp19 gene, and functions in complex with NADPH-ferrihemoprotein reductase in the cytochrome p450 system.Adipose Tissue: Histology deficiency, and congenital adrenal hyperplasiaHyperplasiaAn increase in the number of cells in a tissue or organ without tumor formation. It differs from hypertrophy, which is an increase in bulk without an increase in the number of cells.Cellular Adaptation
DHTDHTA potent androgenic metabolite of testosterone. It is produced by the action of the enzyme 3-oxo-5-alpha-steroid 4-dehydrogenase.Gonadal Hormones:
Follicle stimulating hormone (FSHFSHA major gonadotropin secreted by the adenohypophysis. Follicle-stimulating hormone stimulates gametogenesis and the supporting cells such as the ovarian granulosa cells, the testicular sertoli cells, and leydig cells. Fsh consists of two noncovalently linked subunits, alpha and beta. Within a species, the alpha subunit is common in the three pituitary glycoprotein hormones (TSH, LH, and FSH), but the beta subunit is unique and confers its biological specificity.Menstrual Cycle) and luteinizing hormone (LHLHA major gonadotropin secreted by the adenohypophysis. Luteinizing hormone regulates steroid production by the interstitial cells of the testis and the ovary. The preovulatory luteinizing hormone surge in females induces ovulation, and subsequent luteinization of the follicle. Luteinizing hormone consists of two noncovalently linked subunits, alpha and beta. Within a species, the alpha subunit is common in the three pituitary glycoprotein hormones (TSH, LH, and FSH), but the beta subunit is unique and confers its biological specificity.Menstrual Cycle):
Decrease in Kallmann syndromeKallmann syndromeKallmann syndrome (KS), also called olfacto-genital syndrome, is a genetic condition that causes hypogonadotropic hypogonadism due to decreased secretion of gonadotropin-releasing hormone (GnRH) by the hypothalamus. The lack of sex hormones results in impaired pubertal development. Kallmann Syndrome (due to ↓ gonadotropin-releasing hormoneGonadotropin-releasing hormoneA decapeptide that stimulates the synthesis and secretion of both pituitary gonadotropins, luteinizing hormone and follicle stimulating hormone. Gnrh is produced by neurons in the septum preoptic area of the hypothalamus and released into the pituitary portal blood, leading to stimulation of gonadotrophs in the anterior pituitary gland.Puberty (GnRH))
Increase in 46,XX gonadal dysgenesisGonadal dysgenesisA number of syndromes with defective gonadal developments such as streak gonads and dysgenetic testes or ovaries. The spectrum of gonadal and sexual abnormalities is reflected in their varied sex chromosome (sex chromosomes) constitution as shown by the karyotypes of 45, X monosomy (Turner syndrome); 46, XX (gonadal dysgenesis, 46xx); 46, XY (gonadal dysgenesis, 46, xy); and sex chromosome mosaicism. Their phenotypes range from female, through ambiguous, to male. This concept includes gonadal agenesis.Wilms Tumor and Swyer syndromeSwyer syndromeSwyer syndrome is a disorder of sex development caused by a defect in the SRY gene on chromosome Y. The syndrome is characterized by complete testicular dysgenesis in an individual who has a 46,XY karyotype and is phenotypically female. Swyer Syndrome (normal GnRH function with non-responsive gonadsGonadsThe gamete-producing glands, ovary or testis.Hormones: Overview and Types)
17-Hydroxyprogesterone: ↑ in congenital adrenal hyperplasiaHyperplasiaAn increase in the number of cells in a tissue or organ without tumor formation. It differs from hypertrophy, which is an increase in bulk without an increase in the number of cells.Cellular Adaptation
Imaging
Abdominal pelvic ultrasound to assess the internal organs:
Presence or absence of a uterusUterusThe uterus, cervix, and fallopian tubes are part of the internal female reproductive system. The uterus has a thick wall made of smooth muscle (the myometrium) and an inner mucosal layer (the endometrium). The most inferior portion of the uterus is the cervix, which connects the uterine cavity to the vagina.Uterus, Cervix, and Fallopian Tubes: Anatomy
Consider imaging of the kidneysKidneysThe kidneys are a pair of bean-shaped organs located retroperitoneally against the posterior wall of the abdomen on either side of the spine. As part of the urinary tract, the kidneys are responsible for blood filtration and excretion of water-soluble waste in the urine.Kidneys: Anatomy and urinary tractUrinary tractThe urinary tract is located in the abdomen and pelvis and consists of the kidneys, ureters, urinary bladder, and urethra. The structures permit the excretion of urine from the body. Urine flows from the kidneys through the ureters to the urinary bladder and out through the urethra.Urinary Tract: Anatomy, as abnormalities in the reproductive organs are frequently associated with renal/urinary anomalies.
Principles of Management
Factors affecting management:
Age of presentation (infant, adolescent, versus adult)
PsychotherapyPsychotherapyPsychotherapy is interpersonal treatment based on the understanding of psychological principles and mechanisms of mental disease. The treatment approach is often individualized, depending on the psychiatric condition(s) or circumstance. Psychotherapy/counseling: All patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship (and potentially family members) should be referred for therapy.
Hormone replacement therapyHormone Replacement TherapyHormone replacement therapy (HRT) is used to treat symptoms associated with female menopause and in combination to suppress ovulation. Risks and side effects include uterine bleeding, predisposition to cancer, breast tenderness, hyperpigmentation, migraine headaches, hypertension, bloating, and mood changes.Noncontraceptive Estrogen and Progestins (HRTHRTHormone replacement therapy (HRT) is used to treat symptoms associated with female menopause and in combination to suppress ovulation. Risks and side effects include uterine bleeding, predisposition to cancer, breast tenderness, hyperpigmentation, migraine headaches, hypertension, bloating, and mood changes.Noncontraceptive Estrogen and Progestins) may be indicated in patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship with hormonal deficiencies (e.g., testosteroneTestosteroneA potent androgenic steroid and major product secreted by the leydig cells of the testis. Its production is stimulated by luteinizing hormone from the pituitary gland. In turn, testosterone exerts feedback control of the pituitary LH and FSH secretion. Depending on the tissues, testosterone can be further converted to dihydrotestosterone or estradiol.Androgens and Antiandrogens and/or estrogenEstrogenCompounds that interact with estrogen receptors in target tissues to bring about the effects similar to those of estradiol. Estrogens stimulate the female reproductive organs, and the development of secondary female sex characteristics. Estrogenic chemicals include natural, synthetic, steroidal, or non-steroidal compounds.Ovaries: Anatomy).
Surgery:
Abnormal gonadsGonadsThe gamete-producing glands, ovary or testis.Hormones: Overview and Types, especially gonadoblastomas and dysgerminomas, pose a high risk for malignancyMalignancyHemothorax → surgical excision recommended
Moshiri, M., Chapman, T., Fechner, P.Y., et al. (2012). Evaluation and management of disorders of sex development: Multidisciplinary approach to a complex diagnosis. RadioGraphics. 32(6),1599–1618. https://pubmed.ncbi.nlm.nih.gov/23065160/