


O termo "perturbações do desenvolvimento sexual" refere-se a um grupo de doenças caracterizadas pelo desenvolvimento sexual atípico num indivíduo, que pode envolver anomalias na estrutura e/ou função dos órgãos reprodutores internos e/ou genitália externa. O desenvolvimento sexual típico começa com o sexo cromossómico (por exemplo, 46,XY ou 46,XX), que determina a diferenciação sexual das gónadas (por exemplo, testículos ou ovários), que secretam hormonas que determinam o fenótipo (por exemplo, masculino ou feminino) . A maioria das perturbações do desenvolvimento sexual deve-se a anomalias nos cromossomas, em enzimas ou em recetores específicos. O diagnóstico geralmente envolve a análise do cariótipo e dos níveis hormonais específicos. O tratamento pode ser complexo e geralmente inclui psicoterapia, terapia de reposição hormonal e/ou cirurgia.
Last updated: Dec 15, 2025
As perturbações do desenvolvimento sexual são um grupo de doenças caracterizadas pelo desenvolvimento sexual atípico de um indivíduo, envolvendo anomalias na estrutura e/ou função tanto dos órgãos reprodutores internos quanto da genitália externa.
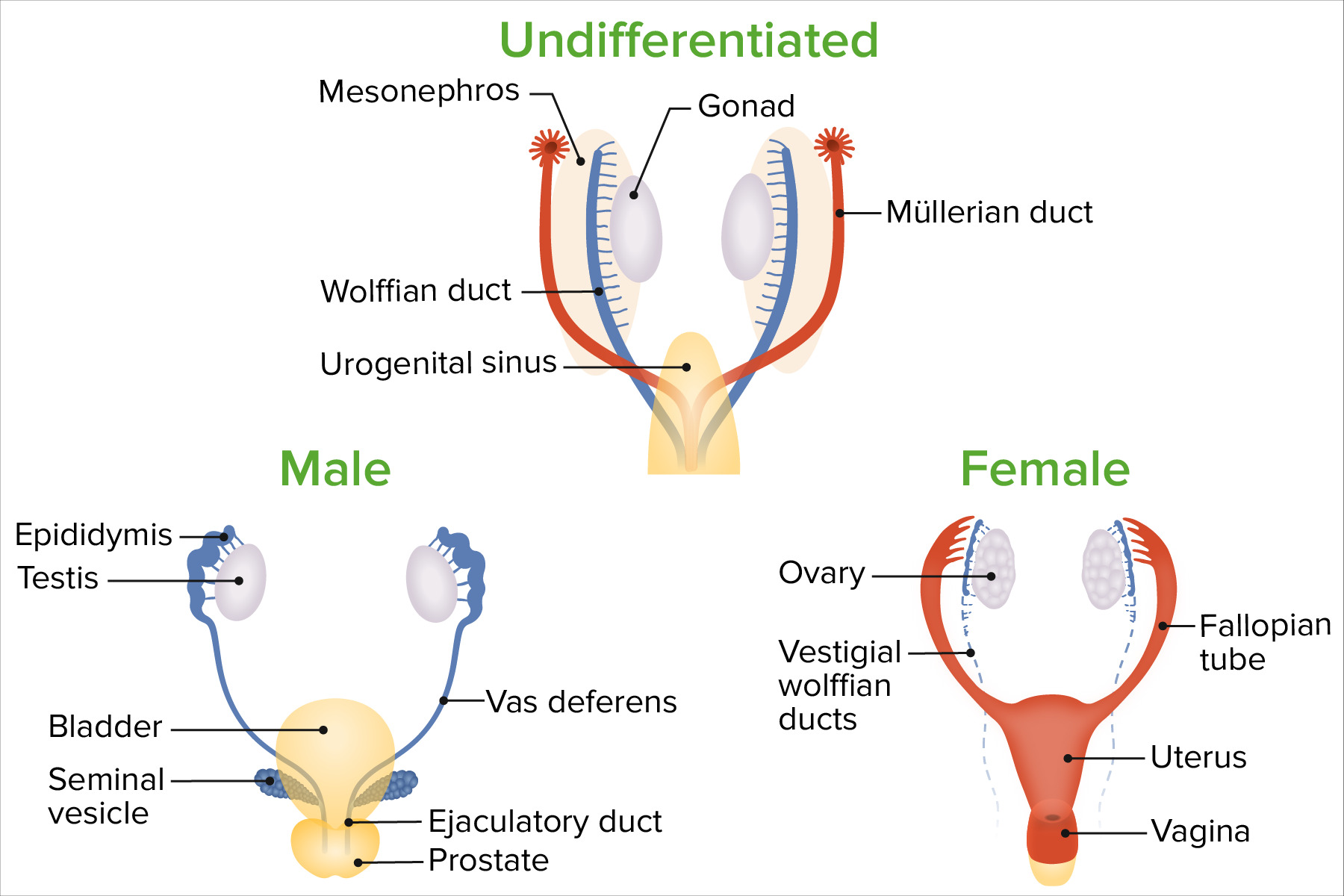
Diferenciação do sexo
Imagem por Lecturio.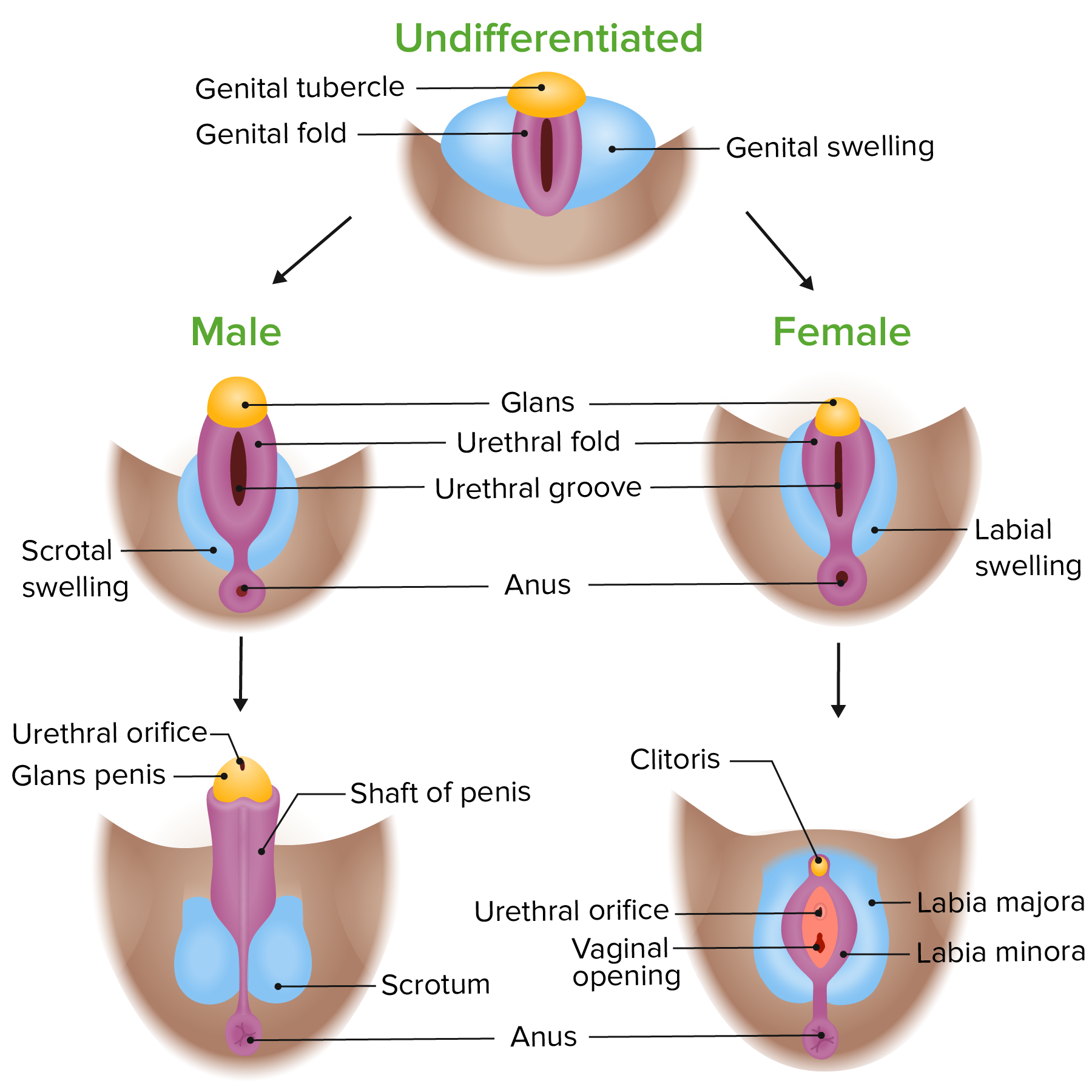
Diferenciação fenotípica da genitália externa em embriões masculinos e femininos
Imagem por Lecturio.O desenvolvimento masculino começa devido à presença da região determinante do sexo do gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics do cromossoma Y ( gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics SRY).
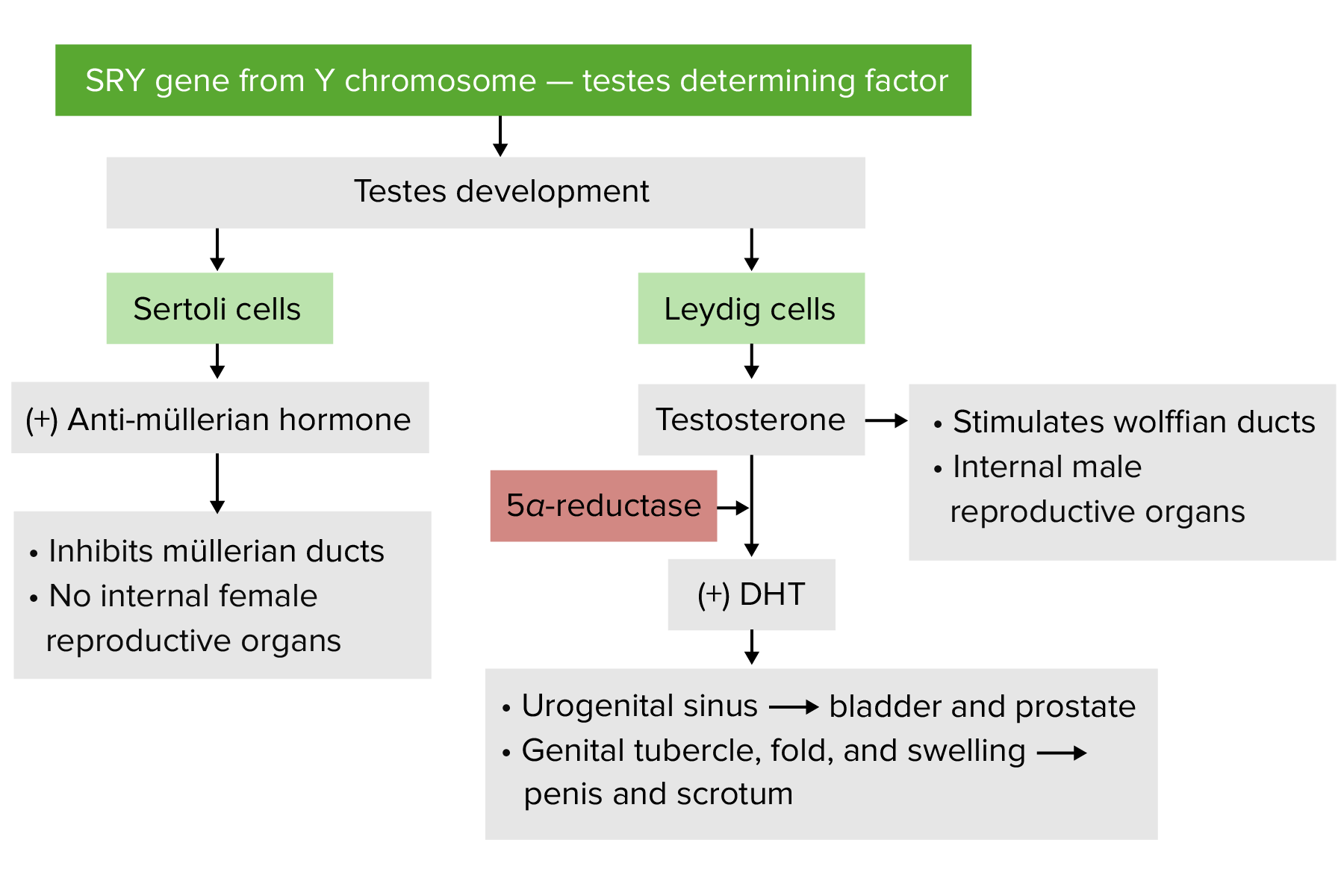
Desenvolvimento masculino
DHT: dihidrotestosterona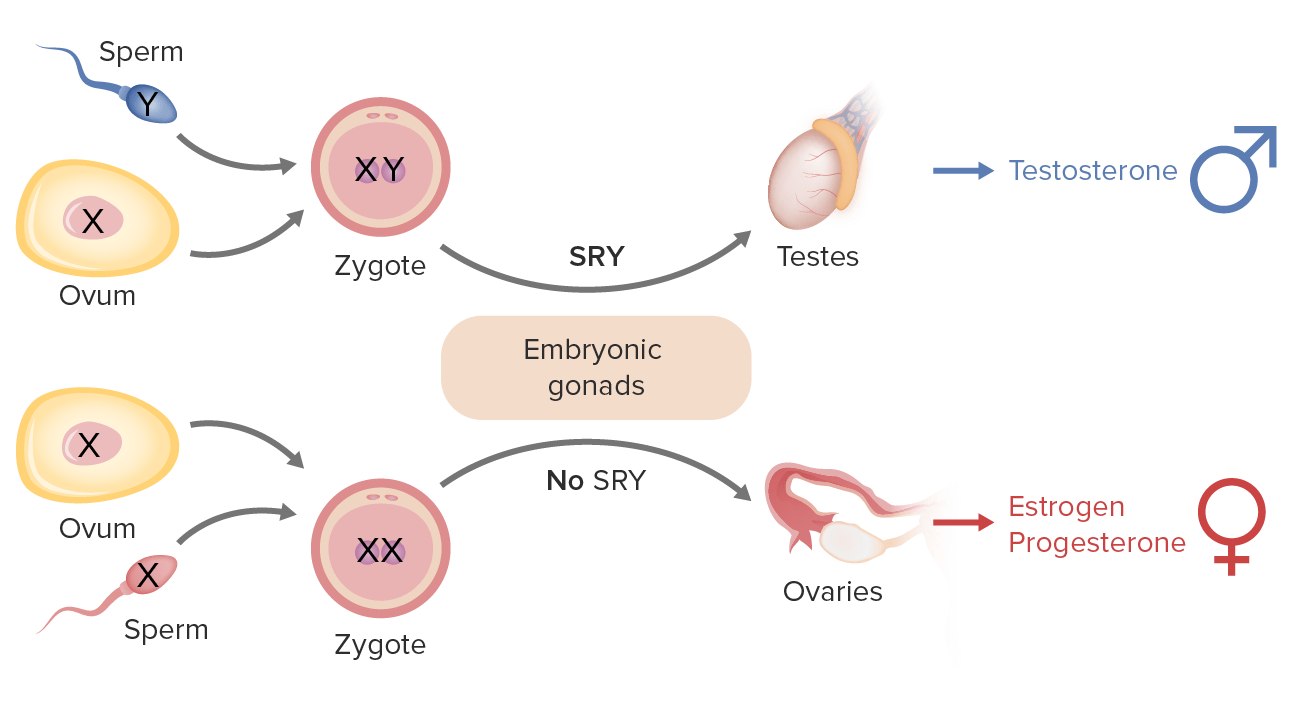
Determinação do sexo em humanos:
O cromossoma Y inclui o gene SRY (sigla em inglês para região determinante do sexo Y) que codifica o fator determinador de testículos (TDF, pela sigla em inglês), que faz com que as gónadas embrionárias se diferenciem em testículos (gónadas masculinas) levando ao desenvolvimento masculino.
Na ausência da proteína TDF (ou seja, sem cromossoma Y), as gónadas embrionárias diferenciam-se em ovários (gónadas femininas).
O sexo feminino tem 2 cópias do cromossoma X (XX);
o sexo masculino tem 1 cromossoma X e 1 cromossoma Y (mais pequeno) (XY).
| Perturbação | Fisiopatologia | Pontos chave |
|---|---|---|
| Síndrome de insensibilidade aos androgénios (SIA) |
|
|
| Défice de 5-α-redutase |
|
|
| Deficiência de aromatase Aromatase An enzyme that catalyzes the desaturation (aromatization) of the ring a of C19 androgens and converts them to C18 estrogens. In this process, the 19-methyl is removed. This enzyme is membrane-bound, located in the endoplasmic reticulum of estrogen-producing cells of ovaries, placenta, testes, adipose, and brain tissues. Aromatase is encoded by the cyp19 gene, and functions in complex with NADPH-ferrihemoprotein reductase in the cytochrome p450 system. Adipose Tissue: Histology |
|
|
| Hiperplasia adrenal congénita |
|
|
| Síndrome de Kallmann |
|
|
| Disgenesia gonadal 46,XX |
|
|
| Síndrome de Swyer |
|
|
| Hermafroditismo verdadeiro |
|
|
| Síndrome de Turner |
|
|
| Síndrome de Klinefelter |
|
|
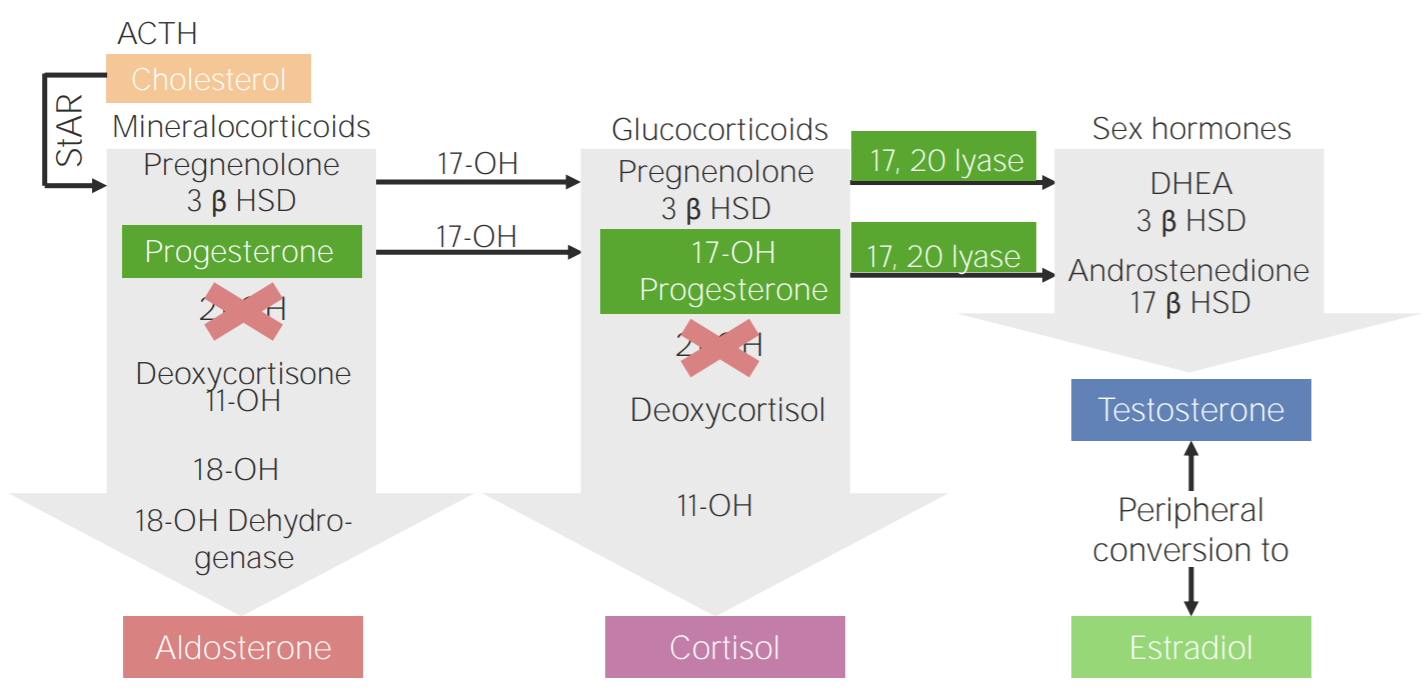
Deficiência de 21-hidroxilase:
Diagrama da fisiopatologia da hiperplasia adrenal congénita devido à deficiência de 21-hidroxilase
O diagnóstico diferencial das perturbações do desenvolvimento sexual geralmente inclui muitas das doenças listadas acima. A abordagem diagnóstica geral para ajudar a determinar o diagnóstico apropriado está listada abaixo.
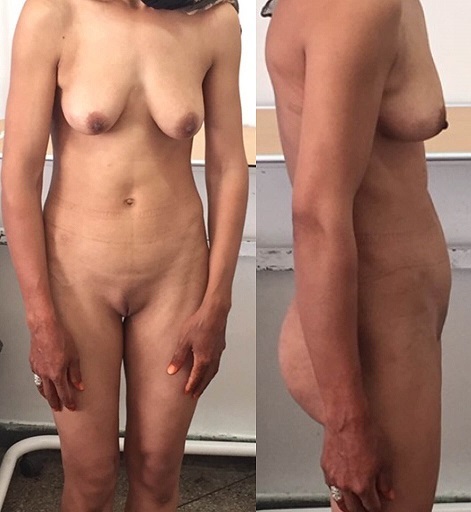
Síndrome de insensibilidade a androgénios:
Paciente de 30 anos com amenorreia primária. Ao exame físico, são notados seios bem desenvolvidos e ausência de pêlos púbicos, indicando exposição ao estrogénio sem exposição significativa à testosterona.
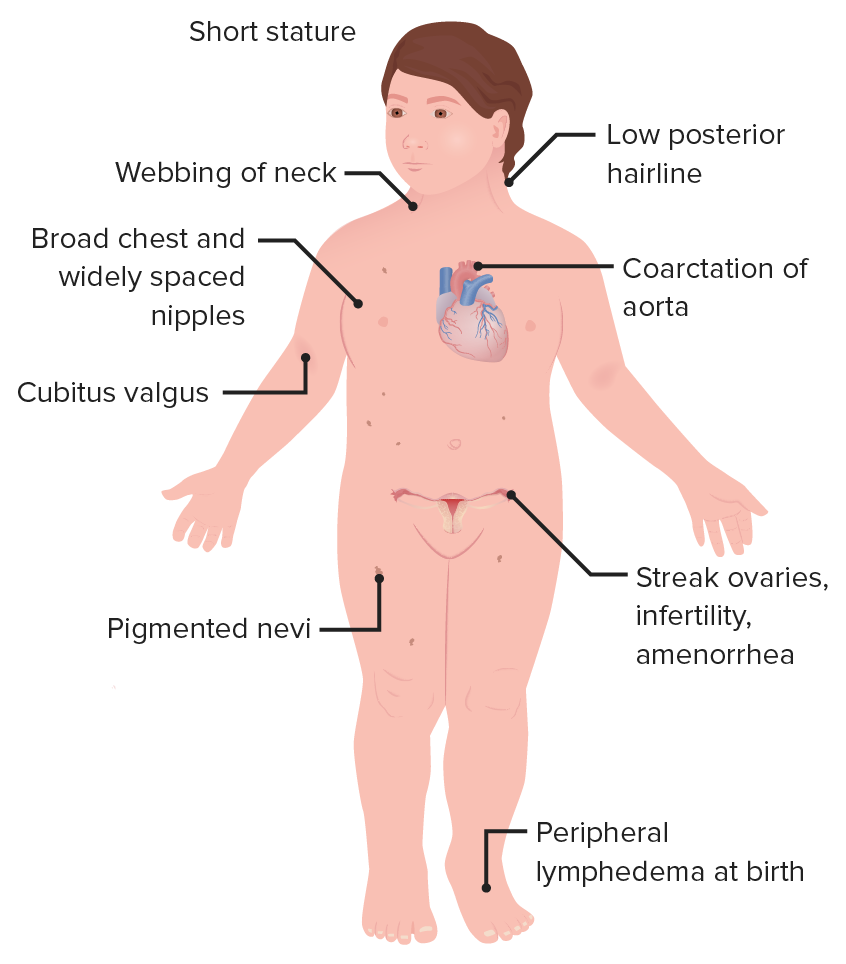
Características de uma mulher com síndrome de Turner
Imagem por Lecturio.Ecografia abdominopélvica para avaliar os órgãos internos:
Considerar a obtenção de imagens dos rins e do trato urinário, porque as anomalias nos órgãos reprodutivos estão frequentemente associadas a anomalias renais/urinárias.
