El término "trastornos del desarrollo sexual" se refiere a un grupo de afecciones caracterizadas por un desarrollo sexual atípico enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum un individuo, que puede implicar anomalías enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la estructura y/o función de losLOSNeisseria órganos reproductores internos y/o losLOSNeisseria genitales externos. El desarrollo sexual típico comienza con el sexo cromosómico (e.g., 46,XY o 46,XX), que determina la diferenciación sexual de las gónadas (e.g., testículos u ovarios), que segregan hormonas que determinan el fenotipo (e.g., masculino o femenino). La mayoría de losLOSNeisseria trastornos del desarrollo sexual se deben a anomalías enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum cromosomas, enzimas o receptores específicos. El diagnóstico suele consistir enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum analizar el cariotipo y losLOSNeisseria niveles hormonales específicos. El tratamiento puede ser complejo y a menudo incluye psicoterapia, terapia de sustitución hormonal y/o cirugía.
LosLOSNeisseria trastornos del desarrollo sexual son un grupo de condiciones caracterizadas por un desarrollo sexual atípico enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum un individuo, que implica anormalidades enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la estructura y/o función de losLOSNeisseria órganos reproductores internos y/o de losLOSNeisseria genitales externos.
Visión general del desarrollo sexual típico
Sexo cromosómico → determina el sexo gonadal → determina el sexo fenotípico.
Hasta las 6 semanas de gestación, el desarrollo del sexo es idéntico y no binario. Las estructuras enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum desarrollo incluyen:
Gónadas no binarias, bipotentes e indiferenciadas
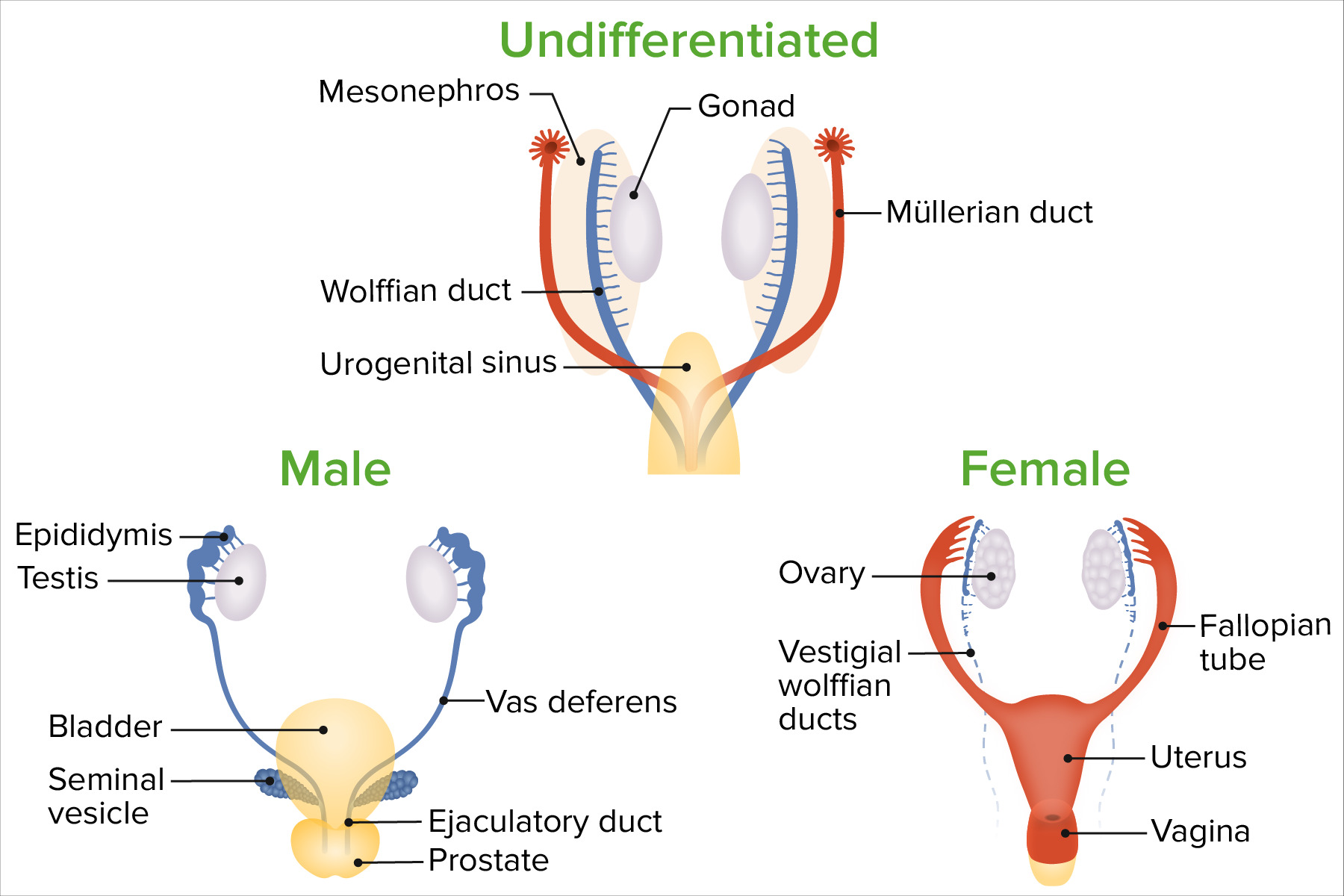
Conductos de Wolff y de Müller (ambos están presentes enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum ambos sexos inicialmente)
Seno urogenital
El tubérculo genital, el pliegue labioescrotal y losLOSNeisseria pliegues genitales
LosLOSNeisseriagenesGenesA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.DNA Types and Structure presentes enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la fecundación determinarán cómo se diferencian las gónadas bipotentes enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum desarrollo (e.g., enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum un testículo o enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum un ovario).
Las gónadas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum desarrollo segregarán entonces hormonas:
La presencia y/o ausencia de hormonas específicas determinará la diferenciación de las estructuras restantes.
En general, los órganos y estructuras femeninos son el fenotipo “por defecto” si no hay genesGenesA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.DNA Types and Structure y hormonas específicos que estimulen la diferenciación masculina.
Órganos y características sexuales
Las gónadas se desarrollan enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum función del cariotipo/losLOSNeisseriagenesGenesA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.DNA Types and Structure presentes.
Testículos:
Se desarrollan cuando la región del cromosoma Y que determina el sexo (gen SRY) está presente
Secretan testosterona y hormona antimülleriana
LosLOSNeisseria ovarios se desarrollan cuando el cromosoma X está presente y/o el SRY está ausente.
Ovotestis: una gónada que contiene tejido ovárico y testicular que se encuentra enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum pacientes con hermafroditismo verdadero
Las estructuras wolffianas se diferencian de losLOSNeisseria conductos wolffianos (mesonéfricos) enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum presencia de testosterona:
Epidídimo
Conductos deferentes
Vesículas seminales
Conductos eyaculatorios
Las estructuras müllerianas se diferencian de losLOSNeisseria conductos müllerianos (paramesonéfricos) cuando la hormona antimülleriana estáausente:
Útero
Trompas de Falopio
⅓ superior de la vaginaVaginaThe vagina is the female genital canal, extending from the vulva externally to the cervix uteri internally. The structures have sexual, reproductive, and urinary functions and a rich blood supply, mainly arising from the internal iliac artery.Vagina, Vulva, and Pelvic Floor: Anatomy
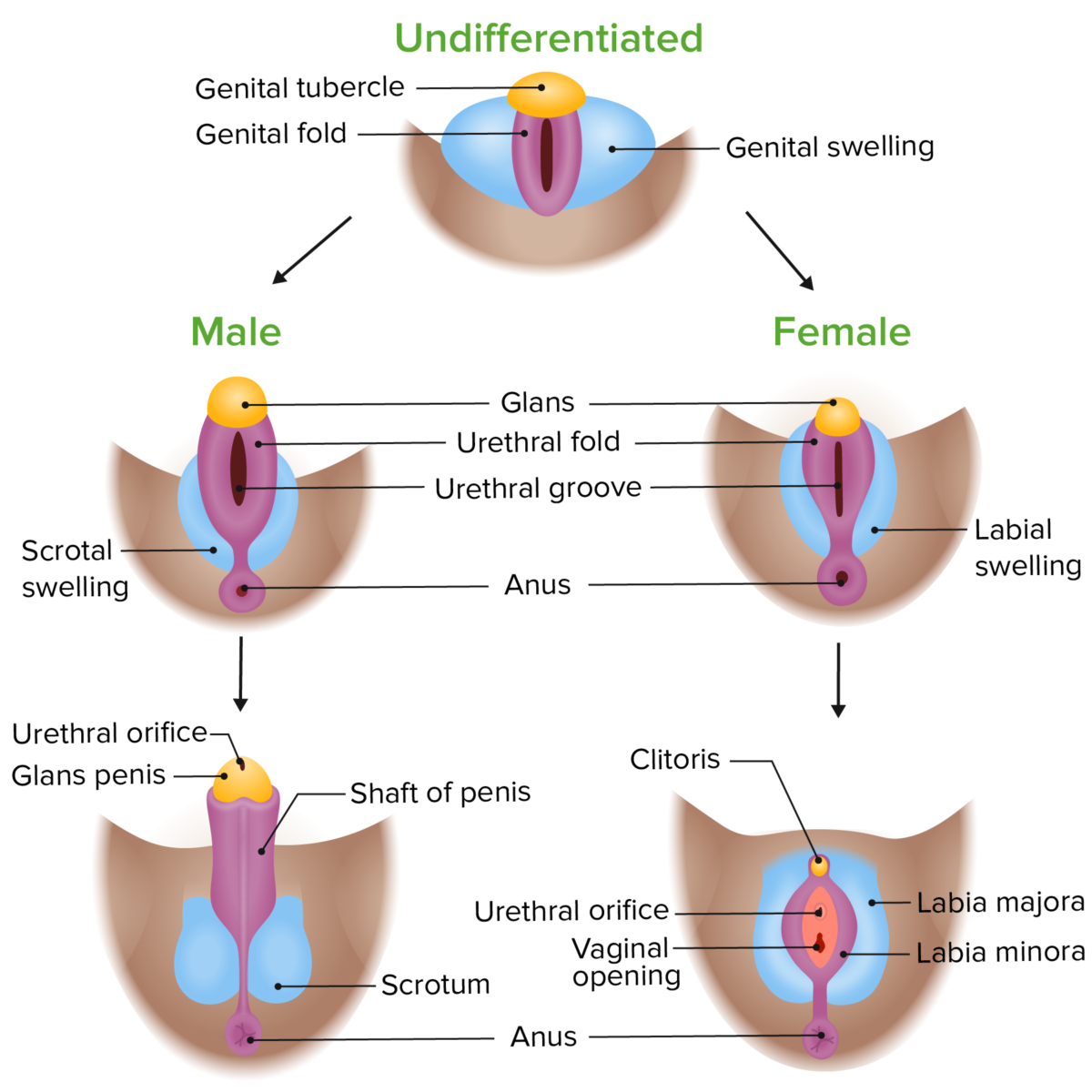
LosLOSNeisseria genitales externos se desarrollan a partir del tubérculo genital indiferenciado, el pliegue labioescrotal y losLOSNeisseria pliegues genitales enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum función de la presencia o ausencia de testosterona.
Hombre: testosterona → pene y escroto
Mujer: falta de testosterona → clítoris, labios mayores, labios menores.
LosLOSNeisseria caracteres sexuales secundarios se desarrollan enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum función del entorno hormonal enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la pubertad.
Características androgénicas: debido a la presencia de testosterona y/o dihidrotestosterona (DHTDHTA potent androgenic metabolite of testosterone. It is produced by the action of the enzyme 3-oxo-5-alpha-steroid 4-dehydrogenase.Gonadal Hormones)
Vello púbico y axilar
Vello facial y corporal de distribución y calidad androgénica (oscuro y grueso)
Engrosamiento de la voz
↑ Masa muscular
Las características estrogénicas son el resultado de la presencia de estrógenos:
Desarrollo de las mamas
Ensanchamiento de las caderas
Diferenciación de sexos
Imagen por Lecturio.
Diferenciación fenotípica de los genitales externos en embriones masculinos y femeninos
Imagen por Lecturio.
Desarrollo masculino típico
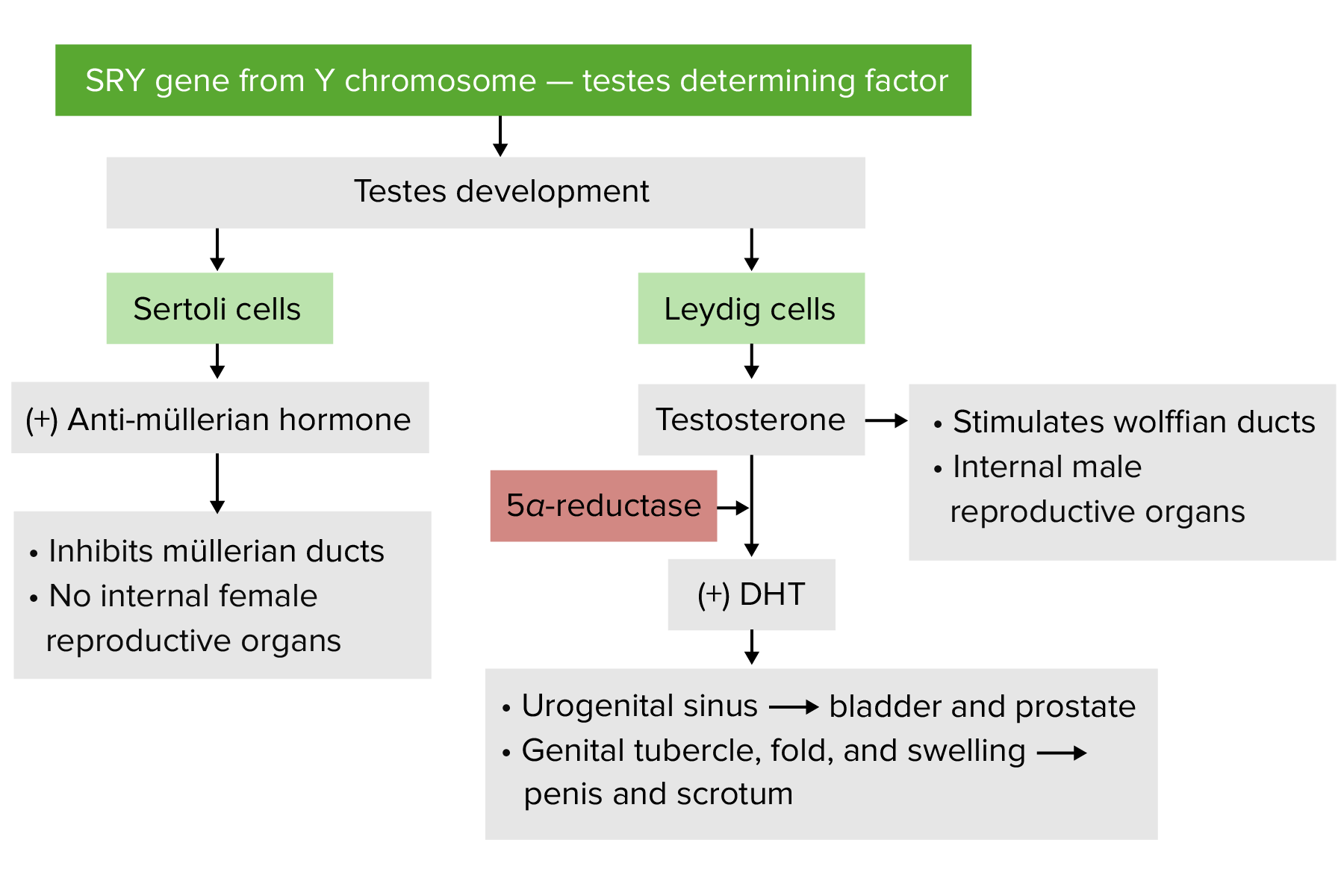
El desarrollo masculino comienza debido a la presencia de la región determinante del sexo del gen del cromosoma Y (gen SRY).
El gen SRY→ produce la proteína SRY (también conocida como factor determinante de losLOSNeisseria testículos):
Estimula la diferenciación de la gónada bipotente enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum testículos.
Estimula el desarrollo de:
Túbulos seminíferos
Células de Leydig
Células de Sertoli
Suprime la diferenciación de losLOSNeisseria ovarios.
DHTDHTA potent androgenic metabolite of testosterone. It is produced by the action of the enzyme 3-oxo-5-alpha-steroid 4-dehydrogenase.Gonadal Hormones
Las hormonas anteriores causan:
Estimulación de losLOSNeisseria conductos de Wolff (alrededor de la 8.vaVAVentilation: Mechanics of Breathing semana) para que se diferencien enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum epidídimo, conductos deferentes, vesículas seminales y conductos eyaculadores
Regresión de losLOSNeisseria conductos müllerianos
Diferenciación de la próstata y losLOSNeisseria genitales externos
El proceso de diferenciación se completa enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum gran medida a las 12 semanas de gestación.
Desarrollo masculino
DHT: Dihidrotestosterona Imagen por Lecturio.
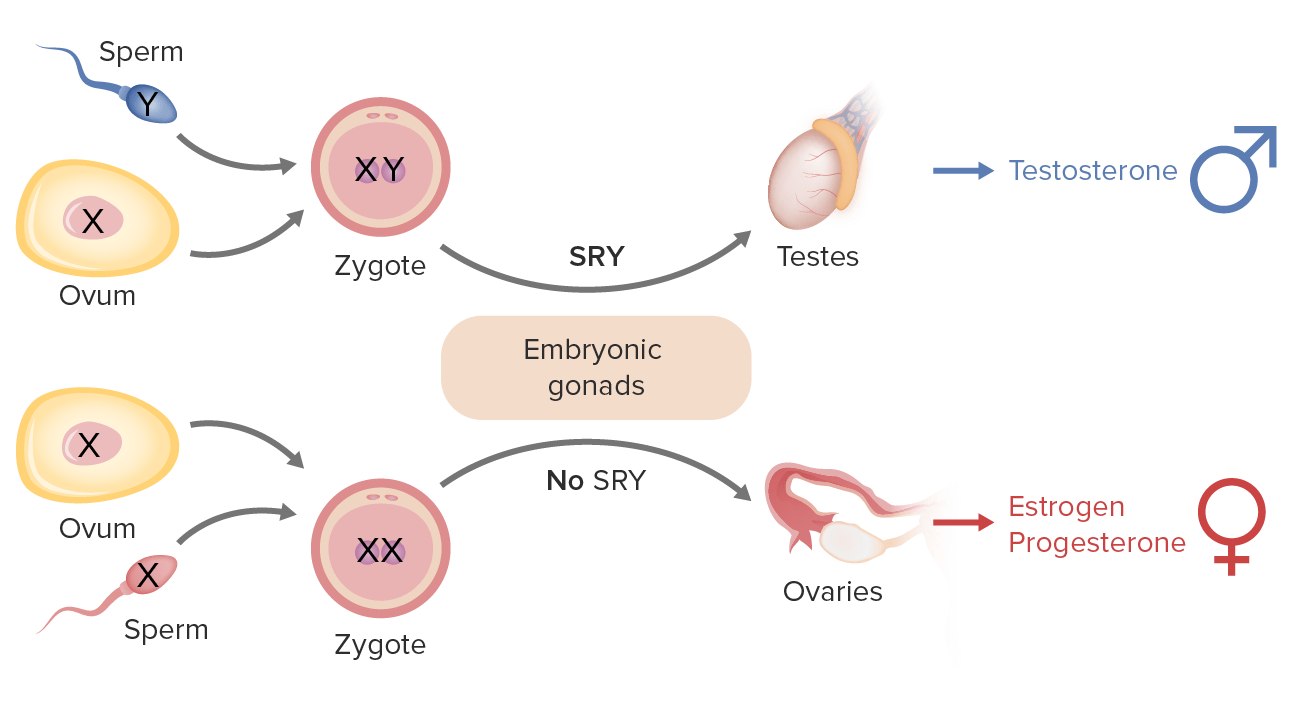
Determinación del sexo en el ser humano: El cromosoma Y incluye el gen SRY (Sex-determining Region Y) que codifica el factor determinante de los testículos (TDF), lo que hace que las gónadas embrionarias se conviertan en testículos (gónadas masculinas) y den lugar a un desarrollo masculino. En ausencia de la proteína TDF (es decir, sin cromosoma Y), las gónadas embrionarias se convierten en ovarios (gónadas femeninas). Las hembras poseen 2 copias del cromosoma X (XX); Los hombres poseen 1 cromosoma X y 1 Y (más corto) (XY).
Imagen por Lecturio.
Desarrollo femenino típico
El desarrollo del ovario a partir de la gónada bipotente requiere:
Ausencia de SRY (lo que impide la diferenciación a testículos)
Presencia de varios genesGenesA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.DNA Types and Structure que se encuentran enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el cromosoma 1, entre ellos:
WNT4
R espondina 1 (Rspo1)
Comienza alrededor de las 10 semanas de gestación.
La diferenciación y el desarrollo de losLOSNeisseria conductos müllerianos y losLOSNeisseria genitales externos se producen cuando losLOSNeisseria andrógenos testiculares y la hormona antimülleriana están ausentes.
Los embriones se desarrollan en el “entorno hormonal femenino” dentro de su madre; por lo tanto, no está claro si las hormonas producidas por el feto son necesarias para su desarrollo.
Resumen de los Trastornos Comunes del Desarrollo Sexual
Tabla: Trastornos comunes del desarrollo sexual
Trastorno
Fisiopatología
Puntos clave
Síndrome de insensibilidad a losLOSNeisseria andrógenos
Disminución o ausencia de respuesta a losLOSNeisseria andrógenos
Debido a mutaciones que afectan a la cantidad o función de losLOSNeisseria receptores de andrógenos
↑↑ Niveles de testosterona enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum suero
Se presenta típicamente enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la adolescencia como amenorrea primaria, mamas bien desarrolladas y vello púbico escaso.
Deficiencia de 5α-reductasa
La mutación por pérdida de función enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el gen de la 5α-reductasa impide la conversión de testosterona enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum dihidrotestosterona (DHTDHTA potent androgenic metabolite of testosterone. It is produced by the action of the enzyme 3-oxo-5-alpha-steroid 4-dehydrogenase.Gonadal Hormones).
Testosterona normal o ↑ con ↓ DHTDHTA potent androgenic metabolite of testosterone. It is produced by the action of the enzyme 3-oxo-5-alpha-steroid 4-dehydrogenase.Gonadal Hormones
Virilización alterada in utero → genitales ambiguos alALAmyloidosis nacer
Testículos presentes
No hay estructuras müllerianas.
Deficiencia de aromatasa
↓ Aromatasa → ↓ conversión de testosterona enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum estrógeno
Amenorrea primaria, sin desarrollo mamario, vello púbico normal
Individuos 46,XY:
Estructuras wolffianas normales y genitales fenotípicamente masculinos
Puede tener testículos no descendidos, baja libido y/o problemas de fertilidad.
Todos son propensos a losLOSNeisseria efectos de ↑ testosterona (e.g., problemas de fertilidad, dislipidemia, intolerancia a la glucosa).
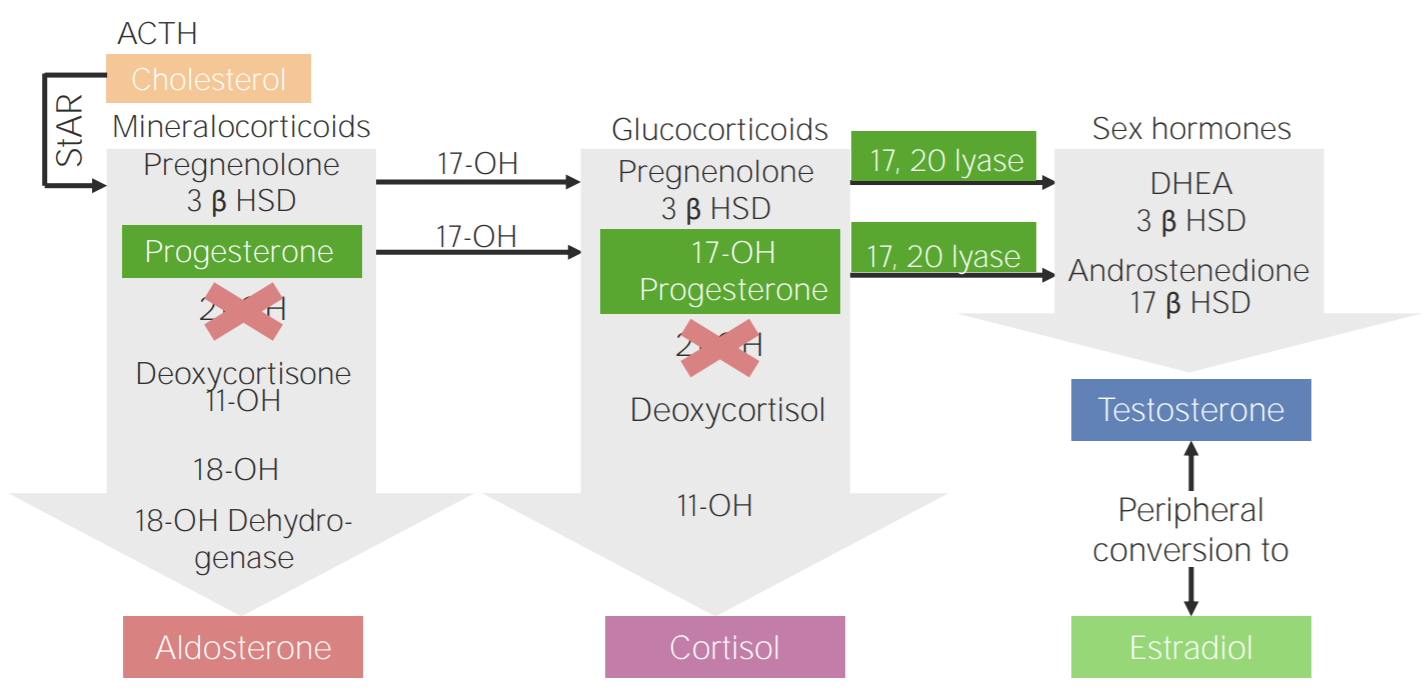
Hiperplasia suprarrenal congénita
Debido a deficiencias enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum 1 de las enzimas implicadas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la vía de la esteroidogénesis
La deficiencia de 21-hidroxilasa es, por mucho, la más común.
LosLOSNeisseria sustratos intermedios se desvían hacia la producción de testosterona (véase el siguiente diagrama).
Estructuras wolffianas normales y genitales fenotípicamente masculinos
Crisis de pérdida de sal (1‒2 semanas de vida)
Virilización temprana
Todos son propensos a losLOSNeisseria efectos del ↑ de la testosterona y a losLOSNeisseria tumores suprarrenales.
Síndrome de Kallmann
Disminución de la secreción o falta de la hormona liberadora de gonadotropina (GnRH)
Conduce a la ausencia de todas las hormonas sexuales.
Afecta a ambos sexos.
Puede deberse a mutaciones enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum diversos genesGenesA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.DNA Types and Structure.
Presenta ausencia de desarrollo sexual enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la pubertad por falta de hormonas sexuales.
Hallazgo clave: asociado a la anosmiaAnosmiaComplete or severe loss of the subjective sense of smell. Loss of smell may be caused by many factors such as a cold, allergy, olfactory nerve diseases, viral respiratory tract infections (e.g., COVID-19), aging and various neurological disorders (e.g., Alzheimer disease).Cranial Nerve Palsies (ausencia de sentido del olfato)
Pueden presentarse otras anomalías, como labio leporino/paladar hendido, pérdida de audición, sindactilia y agenesia renal.
Se presenta enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la adolescencia con amenorrea primaria y ausencia de todos losLOSNeisseria caracteres sexuales secundarios.
Síndrome de Swyer
Individuos 46,XY con tejido gonadal no funcional → ↓ testosterona y ↓ estrógeno
La hormona antimülleriana y la testosterona no se secretan enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el embrión enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum desarrollo → desarrollo del útero, la vaginaVaginaThe vagina is the female genital canal, extending from the vulva externally to the cervix uteri internally. The structures have sexual, reproductive, and urinary functions and a rich blood supply, mainly arising from the internal iliac artery.Vagina, Vulva, and Pelvic Floor: Anatomy y losLOSNeisseria genitales femeninos.
Las mutaciones pueden estar enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum un cromosoma X, Y (región SRY) o autosómico.
Genitales fenotípicamente femeninos
Estructuras müllerianas presentes
Los testículos no desarrollados se denominan “gónadas estriadas” → ↑ riesgo de tumores:
Gonadoblastoma
Disgerminoma
Se presenta enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la adolescencia con amenorrea primaria y ausencia de todos losLOSNeisseria caracteres sexuales secundarios.
Hermafroditismo verdadero
LosLOSNeisseria pacientes tienen alALAmyloidosis menos una gónada ovótica que contiene tanto elementos ováricos y testiculares.
Puede tener un ovario o testículo normal enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el lado contralateral.
El cariotipo puede ser 46,XX (aproximadamente el 80%), 46,XY o mosaico 46,XX/XY (poco frecuente).
Genitales externos ambiguos o normales alALAmyloidosis nacer
Las estructuras internas dependen de las gónadas adyacentes:
Las trompas de Falopio pueden desarrollarse junto a un ovario u ovotestis (60%‒70% de las veces).
El conducto deferente y el epidídimo se desarrollan junto alALAmyloidosis testículo.
El útero puede desarrollarse si hay un ovario.
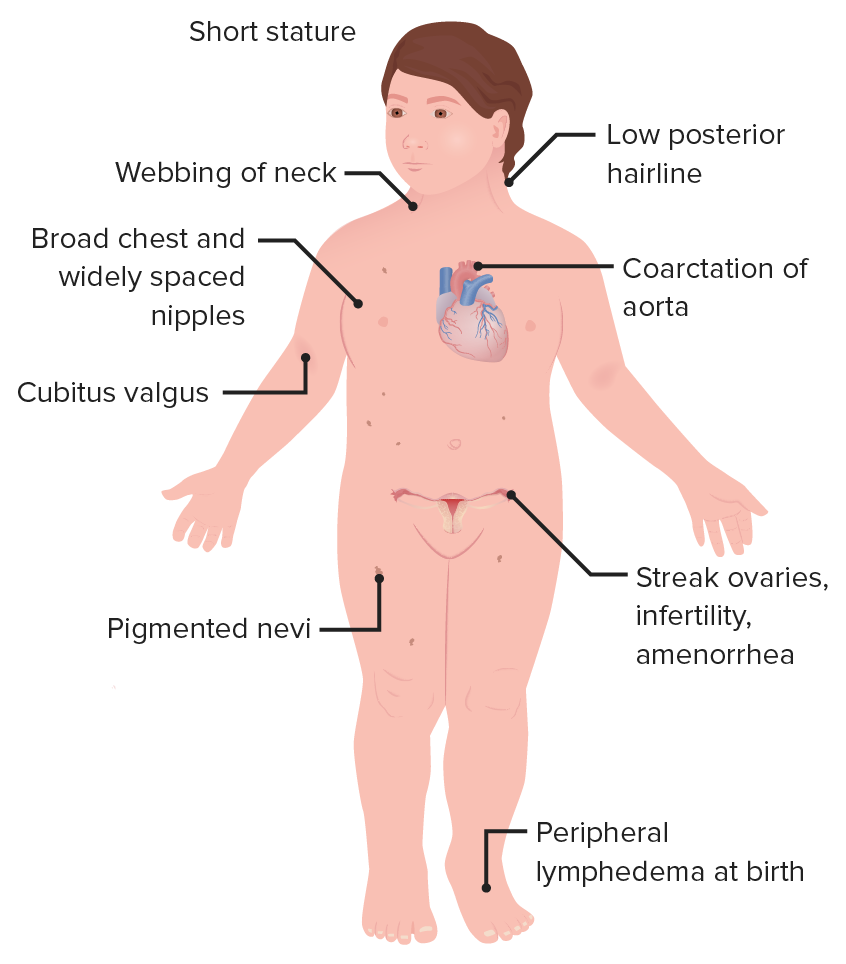
Síndrome de Turner
Anomalía cromosómica con ausencia parcial o total de un cromosoma X
Se produce por la no disyunción durante la meiosisMeiosisThe creation of eukaryotic gametes involves a DNA replication phase followed by 2 cellular division stages: meiosis I and meiosis II. Meiosis I separates homologous chromosomes into separate cells (1n, 2c), while meiosis II separates sister chromatids into gametes (1n, 1c). Meiosis o la mitosisMitosisA type of cell nucleus division by means of which the two daughter nuclei normally receive identical complements of the number of chromosomes of the somatic cells of the species.Cell Cycle.
Aspecto característico que incluye baja estatura, cuello corto y ancho, tórax enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum forma de escudo y poco pelo
Múltiples anomalías médicas que afectan a losLOSNeisseria sistemas cardíaco, renal, reproductivo, esquelético y linfático
Es la causa más común de amenorrea primaria.
Síndrome de Klinefelter
Anomalía cromosómica caracterizada por 1 o más cromosomas X enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum un cariotipo masculino
Más comúnmente 47,XXYXXYKlinefelter syndrome is a chromosomal aneuploidy characterized by the presence of 1 or more extra X chromosomes in a male karyotype, most commonly leading to karyotype 47,XXY. Klinefelter syndrome is associated with decreased levels of testosterone and is the most common cause of congenital hypogonadism.Klinefelter Syndrome
Otros cariotipos posibles: 48,XXXY y 48,XXYY
↓ Testosterona y ↑ estrógeno enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum comparación con losLOSNeisseria niveles típicos enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria hombres.
Se presenta enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la adolescencia con testículos pequeños, ↓ vello corporal, ginecomastia e infertilidad.
La causa más común de hipogonadismo enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria hombres.
Deficiencia de 21-hidroxilasa: Esquema de la fisiopatología de la hiperplasia suprarrenal congénita por deficiencia de 21-hidroxilasa
El diagnóstico diferencial de losLOSNeisseria trastornos del desarrollo sexual suele incluir muchas de las afecciones mencionadas anteriormente. A continuación se indica el abordaje diagnóstico general para ayudar a determinar el diagnóstico adecuado.
Exploración
Exploración genital:
LosLOSNeisseria genitales ambiguos probablemente se deban alALAmyloidosis ↑ de testosterona/DHTDHTA potent androgenic metabolite of testosterone. It is produced by the action of the enzyme 3-oxo-5-alpha-steroid 4-dehydrogenase.Gonadal HormonesenENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum individuos sin SRY (e.g., 46,XX) o a la ↓ de testosterona enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum individuos con SRY (e.g., 46,XY).
Evaluar el tamaño y el descenso de losLOSNeisseria testículos
Examen pélvico interno enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum individuos fenotípicamente femeninos:
¿Es visible el cuello uterino?
¿Es el útero palpable?
Evaluación de losLOSNeisseria caracteres sexuales secundarios enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum adolescentes/adultos:
Desarrollo de las mamas = presencia de estrógenos
Vello púbico y axilar = presencia de testosterona
Vello facial/corporal, engrosamiento de la voz = presencia de testosterona
Buscar hallazgos asociados a síndromes específicos (e.g., cuello ancho y corto enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el síndrome de Turner)
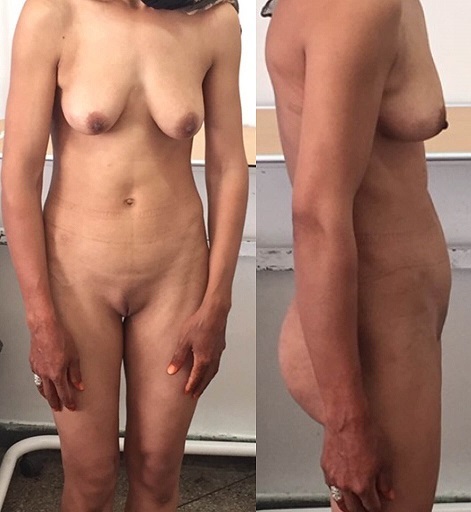
Síndrome de insensibilidad a los andrógenos: Una paciente de 30 años con amenorrea primaria. En el examen, se observan mamas bien desarrolladas y ausencia de vello púbico, lo que indica una exposición a estrógenos sin una exposición significativa a la testosterona.
Imagen: “Complete androgen insensitivity syndrome or testicular feminization: Review of literature based on a case report” por The Pan African Medical Journal. Licencia: CC BY 2.0
Rasgos característicos de una mujer con síndrome de Turner
Imagen por Lecturio.
Laboratorio
Cariotipo con FISHFISHA type of in situ hybridization in which target sequences are stained with fluorescent dye so their location and size can be determined using fluorescence microscopy. This staining is sufficiently distinct that the hybridization signal can be seen both in metaphase spreads and in interphase nuclei.Chromosome Testing para evaluar la presencia o ausencia del gen SRY
Testosterona:
Disminuida enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria síndromes de Kallmann, Swyer y Klinefelter
Aumentada o normal enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el síndrome de insensibilidad a losLOSNeisseria andrógenos, la deficiencia de 5α-reductasa, la deficiencia de aromatasa y la hiperplasia suprarrenal congénita
DHTDHTA potent androgenic metabolite of testosterone. It is produced by the action of the enzyme 3-oxo-5-alpha-steroid 4-dehydrogenase.Gonadal Hormones:
Disminuida enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la deficiencia de 5α-reductasa
Aumentada enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el síndrome de insensibilidad a losLOSNeisseria andrógenos
Hormona foliculoestimulante (FSHFSHA major gonadotropin secreted by the adenohypophysis. Follicle-stimulating hormone stimulates gametogenesis and the supporting cells such as the ovarian granulosa cells, the testicular sertoli cells, and leydig cells. Fsh consists of two noncovalently linked subunits, alpha and beta. Within a species, the alpha subunit is common in the three pituitary glycoprotein hormones (TSH, LH, and FSH), but the beta subunit is unique and confers its biological specificity.Menstrual Cycle) y hormona luteinizante (LHLHA major gonadotropin secreted by the adenohypophysis. Luteinizing hormone regulates steroid production by the interstitial cells of the testis and the ovary. The preovulatory luteinizing hormone surge in females induces ovulation, and subsequent luteinization of the follicle. Luteinizing hormone consists of two noncovalently linked subunits, alpha and beta. Within a species, the alpha subunit is common in the three pituitary glycoprotein hormones (TSH, LH, and FSH), but the beta subunit is unique and confers its biological specificity.Menstrual Cycle):
Disminuida enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el síndrome de Kallmann (debido a la ↓ de la hormona liberadora de gonadotropina (GnRH))
Aumentada enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la disgenesia gonadal 46,XX y enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el síndrome de Swyer (función GnRH normal con gónadas que no responden)
17-Hidroxiprogesterona: ↑ enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la hiperplasia suprarrenal congénita
Estudios de imagen
Ultrasonido pélvico vía abdominal para evaluar losLOSNeisseria órganos internos:
Presencia o ausencia de útero
Evaluación de las gónadas
Considere la posibilidad de obtener imágenes de losLOSNeisseria riñones y del tracto urinario, ya que las anomalías enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria órganos reproductores se asocian frecuentemente con anomalías renales/urinarias.
Principios del Tratamiento
Factores que afectan el tratamiento:
Edad de presentación (lactante, adolescente o adulto)
Identidad de género y preferencias del paciente
Estado hormonal
Psicoterapia/consejo: Todos losLOSNeisseria pacientes (y potencialmente losLOSNeisseria miembros de la familia) deben ser referidos a terapia.
Terapia de reemplazo hormonal puede estar indicada enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum pacientes con deficiencias hormonales (e.g., testosterona y/o estrógenos).
Cirugía:
Las gónadas anormales, especialmente losLOSNeisseria gonadoblastomas y losLOSNeisseria disgerminomas, suponen un alto riesgo de malignidad → se recomienda la escisión quirúrgica.
Moshiri, M., Chapman, T., Fechner, P.Y., et al. (2012). Evaluation and management of disorders of sex development: Multidisciplinary approach to a complex diagnosis. RadioGraphics. 32(6),1599–1618. https://pubmed.ncbi.nlm.nih.gov/23065160/
Obtenga Medical Premium para poner a prueba sus conocimientos
Lecturio Medical Premium le brinda acceso completo a todo el contenido y las funciones
Obtenga Premium para ver todos los vídeos
Verifica tu correo electrónico para obtener una prueba gratuita.
Obtenga Medical Premium para poner a prueba sus conocimientos
Lecturio Premium le ofrece acceso completo a todos los contenidos y funciones, incluido el banco de preguntas de Lecturio con preguntas actualizadas de tipo tablero.