Sturge-Weber syndrome (SWS) is a congenital neurocutaneous disorder presenting with a facial birthmark called a port-wine stain (PWS), neurological abnormalities such as seizures Seizures A seizure is abnormal electrical activity of the neurons in the cerebral cortex that can manifest in numerous ways depending on the region of the brain affected. Seizures consist of a sudden imbalance that occurs between the excitatory and inhibitory signals in cortical neurons, creating a net excitation. The 2 major classes of seizures are focal and generalized. Seizures, and eye abnormalities such as glaucoma Glaucoma Glaucoma is an optic neuropathy characterized by typical visual field defects and optic nerve atrophy seen as optic disc cupping on examination. The acute form of glaucoma is a medical emergency. Glaucoma is often, but not always, caused by increased intraocular pressure (IOP). Glaucoma. Not all of these symptoms have to be present in an affected individual, and some may develop later in life. While the condition is congenital, it is not inherited, as the causative mutation Mutation Genetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations in the gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics GNAQ is somatic and sporadic Sporadic Selective IgA Deficiency. Diagnosis is suspected based on symptoms and neuroimaging Neuroimaging Non-invasive methods of visualizing the central nervous system, especially the brain, by various imaging modalities. Febrile Infant, and confirmed with genetic testing Genetic Testing Detection of a mutation; genotype; karyotype; or specific alleles associated with genetic traits, heritable diseases, or predisposition to a disease, or that may lead to the disease in descendants. It includes prenatal genetic testing. Myotonic Dystrophies. Management is targeted at symptom management and prevention of seizures Seizures A seizure is abnormal electrical activity of the neurons in the cerebral cortex that can manifest in numerous ways depending on the region of the brain affected. Seizures consist of a sudden imbalance that occurs between the excitatory and inhibitory signals in cortical neurons, creating a net excitation. The 2 major classes of seizures are focal and generalized. Seizures and hemiparesis Hemiparesis The term hemiparesis refers to mild to moderate weakness involving one side of the body. Epidural Hemorrhage.
Last updated: Dec 15, 2025
The incidence Incidence The number of new cases of a given disease during a given period in a specified population. It also is used for the rate at which new events occur in a defined population. It is differentiated from prevalence, which refers to all cases in the population at a given time. Measures of Disease Frequency is approximately 1 in 20,000 to 50,000 live newborns.
Symptoms are usually due to the effect of capillary-venous malformation within the brain Brain The part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem. Nervous System: Anatomy, Structure, and Classification, skin Skin The skin, also referred to as the integumentary system, is the largest organ of the body. The skin is primarily composed of the epidermis (outer layer) and dermis (deep layer). The epidermis is primarily composed of keratinocytes that undergo rapid turnover, while the dermis contains dense layers of connective tissue. Skin: Structure and Functions, and eye. Not all symptoms have to be present in an individual for them to be diagnosed with SWS, and some symptoms may develop later in life.
Port-wine stain (PWS):
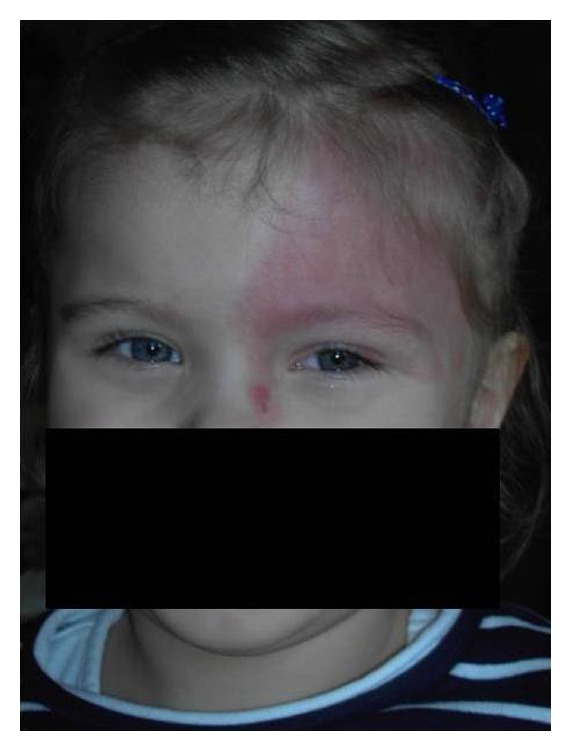
Port-wine stain (PWS):
Diffuse nevus flammeus involving the left upper eyelid and forehead
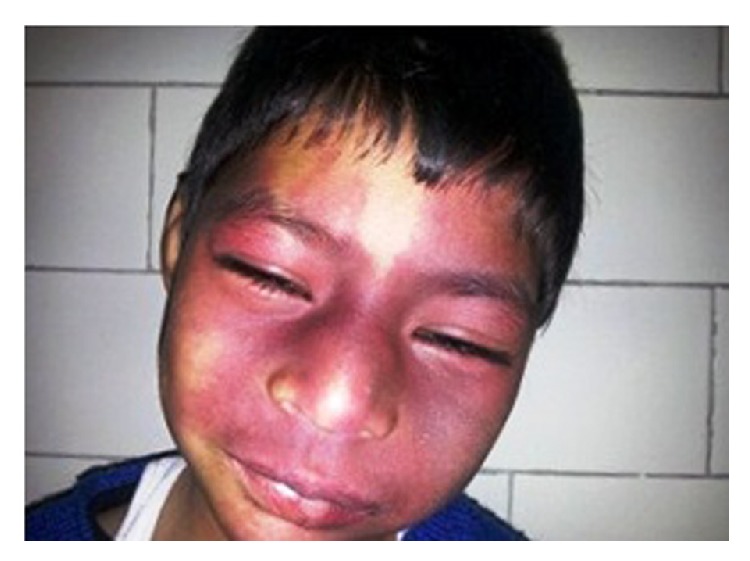
Port-wine stain (PWS):
Bilateral nevus flammeus over the face
An easy way to remember is by using the mnemonic STURGE:
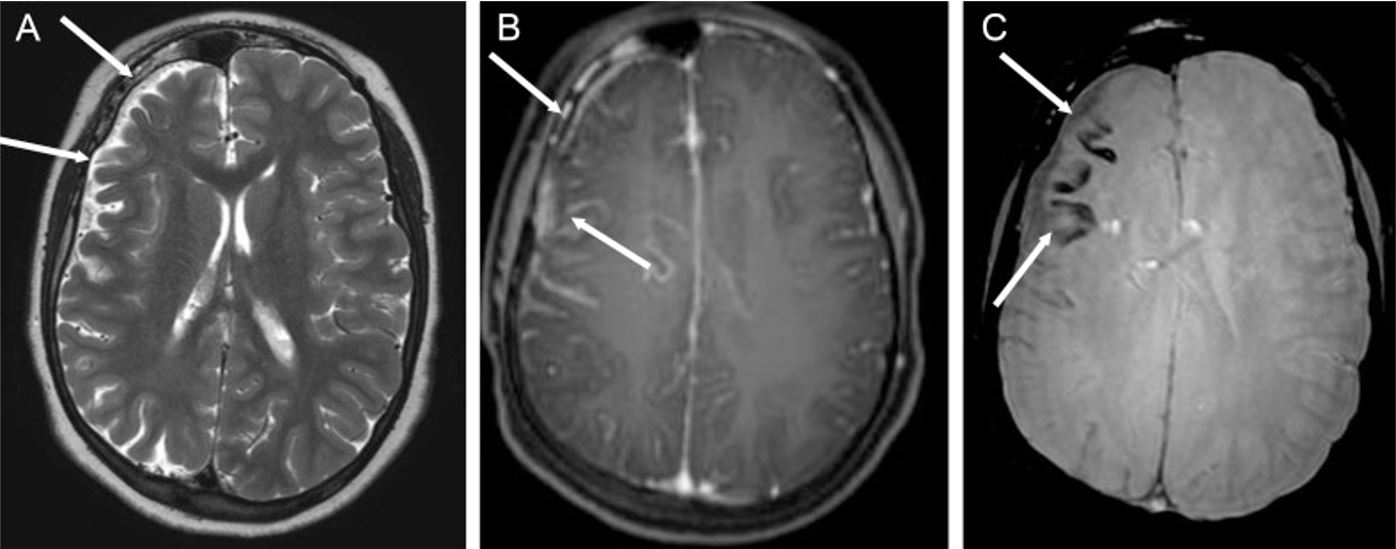
Epilepsy associated with Sturge-Weber syndrome (SWS): Sturge-Weber syndrome in a 14-year-old boy with multiple epileptic foci in the right cerebral hemisphere and chronic partial epilepsy
A: axial T2-weighted image showing right cerebral hemiatrophy (white arrows)
B: axial T1-weighted sequence after gadolinium injection, demonstrating leptomeningeal enhancement of the frontoparietal region (white arrows)
C: White arrows point to cortical calcification in the frontal lobe, appearing as signal voids from subjected weighted images.