Tuberous sclerosis Sclerosis A pathological process consisting of hardening or fibrosis of an anatomical structure, often a vessel or a nerve. Wilms Tumor or tuberous sclerosis Sclerosis A pathological process consisting of hardening or fibrosis of an anatomical structure, often a vessel or a nerve. Wilms Tumor complex (TSC) is an autosomal dominant Autosomal dominant Autosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal dominant diseases are expressed when only 1 copy of the dominant allele is inherited. Autosomal Recessive and Autosomal Dominant Inheritance disorder with mainly neurocutaneous symptoms. Mutation Mutation Genetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations in the TSC genes Genes A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. DNA Types and Structure causes excessive tumor-like growths in the brain Brain The part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem. Nervous System: Anatomy, Structure, and Classification, eyes, heart, kidney, and lungs Lungs Lungs are the main organs of the respiratory system. Lungs are paired viscera located in the thoracic cavity and are composed of spongy tissue. The primary function of the lungs is to oxygenate blood and eliminate CO2. Lungs: Anatomy. Cutaneous manifestations include hypopigmentation Hypopigmentation A condition caused by a deficiency or a loss of melanin pigmentation in the epidermis, also known as hypomelanosis. Hypopigmentation can be localized or generalized, and may result from genetic defects, trauma, inflammation, or infections. Malassezia Fungi (i.e., ash leaf spots, confetti lesions) or excessive growth (i.e., angiofibroma, shagreen patch Patch Nonpalpable lesion > 1 cm in diameter Generalized and Localized Rashes). The diagnosis is made on clinical suspicion and confirmed by genetic testing Genetic Testing Detection of a mutation; genotype; karyotype; or specific alleles associated with genetic traits, heritable diseases, or predisposition to a disease, or that may lead to the disease in descendants. It includes prenatal genetic testing. Myotonic Dystrophies. Management entails a multidisciplinary approach that targets monitoring and treatment of the various manifestations of the disorder. mTOR mTOR Peutz-Jeghers Syndrome inhibitors such as sirolimus Sirolimus A macrolide compound obtained from streptomyces hygroscopicus that acts by selectively blocking the transcriptional activation of cytokines thereby inhibiting cytokine production. It is bioactive only when bound to immunophilins. Sirolimus is a potent immunosuppressant and possesses both antifungal and antineoplastic properties. Immunosuppressants and everolimus Everolimus A derivative of sirolimus and an inhibitor of tor serine-threonine kinases. It is used to prevent graft rejection in heart and kidney transplant patients by blocking cell proliferation signals. It is also an antineoplastic agent. Immunosuppressants are used to treat severe manifestations.
Last updated: Dec 15, 2025
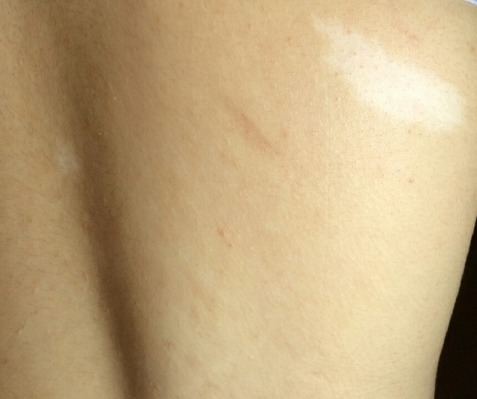
Hypopigmented macule (ash leaf spot)
Image: “Hypomelanotic lesions (ash leaf spot) on skin” by Falsafi P, Taghavi-Zenouz A, Khorshidi-Khiyavi R, Nezami N, Estiar MA. License: CC BY 3.0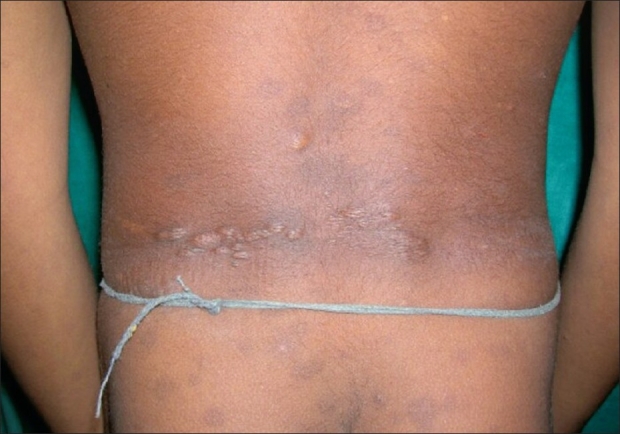
Shagreen patch noted in the lower back
Image: “Shagreen patches on the back in a child” by Ghosh SK, Bandyopadhyay D, Chatterjee G, Ghosh A, Sarkar S, Sarkar S. License: CC BY 2.0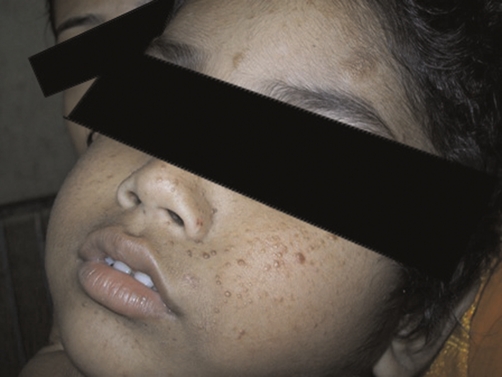
Angiofibroma
Image: “Shows the characteristics skin lesions on the face” by Datta AK, Mandal S, Bhattacharya S. License: CC BY 3.0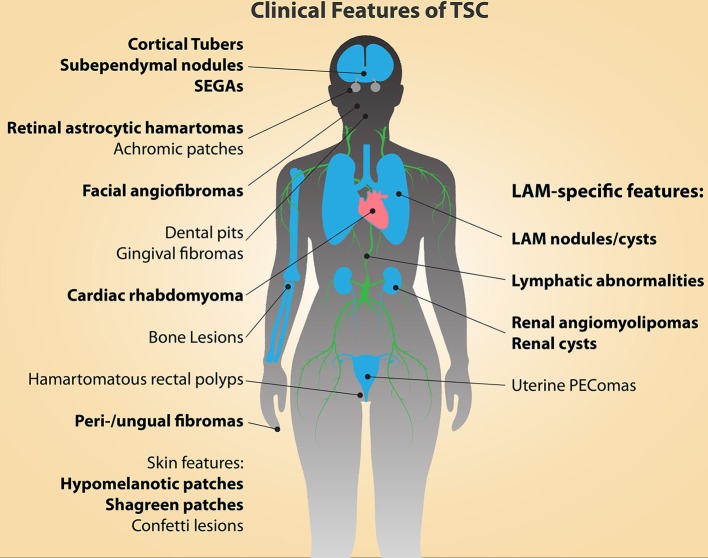
Major diagnostic features of tuberous sclerosis complex (TSC; indicated in bold type)
LAM: lymphangioleiomyomatosis
PEComa: tumor showing perivascular epithelioid cell differentiation
SEGA: subependymal giant-cell astrocytoma
No single clinical presentation of TSC is considered diagnostic.
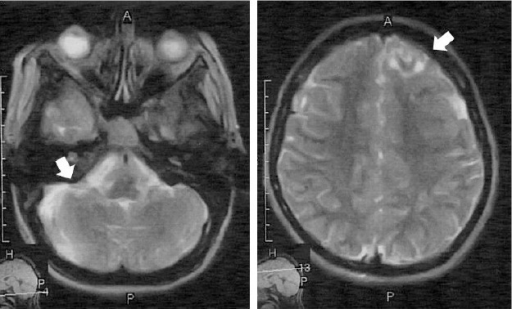
MRI showing cortical and subcortical tubers
Image: “F5” by Parisa Falsafi et al. License: CC BY 3.0