Inclusion-cell disease (I-cell disease, mucolipidosis II, or ML II) is caused by a defect in uridine diphosphate Uridine diphosphate A uracil nucleotide containing a pyrophosphate group esterified to C5 of the sugar moiety. Purine and Pyrimidine Metabolism (UDP)-N-acetylgucosamine-1-phosphotransferase, an enzyme that transfers phosphate Phosphate Inorganic salts of phosphoric acid. Electrolytes to mannose residues on specific proteins Proteins Linear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein. Energy Homeostasis. Lysosomes Lysosomes A class of morphologically heterogeneous cytoplasmic particles in animal and plant tissues characterized by their content of hydrolytic enzymes and the structure-linked latency of these enzymes. The intracellular functions of lysosomes depend on their lytic potential. The single unit membrane of the lysosome acts as a barrier between the enzymes enclosed in the lysosome and the external substrate. The activity of the enzymes contained in lysosomes is limited or nil unless the vesicle in which they are enclosed is ruptured or undergoes membrane fusion. The Cell: Organelles must have this protein, since it is responsible for the breakdown of oligosaccharides Oligosaccharides Carbohydrates consisting of between two (disaccharides) and ten monosaccharides connected by either an alpha- or beta-glycosidic link. They are found throughout nature in both the free and bound form. Basics of Carbohydrates, lipids Lipids Lipids are a diverse group of hydrophobic organic molecules, which include fats, oils, sterols, and waxes. Fatty Acids and Lipids, and glycosaminoglycans. A deficiency in this enzyme results in the accumulation of these chemicals in the body, causing “inclusion cells.” Patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship present within the 1st year of life with failure to thrive Failure to Thrive Failure to thrive (FTT), or faltering growth, describes suboptimal weight gain and growth in children. The majority of cases are due to inadequate caloric intake; however, genetic, infectious, and oncological etiologies are also common. Failure to Thrive and developmental delay. Blood testing for enzyme activity and the presence of inclusion bodies are diagnostic. Treatment is aimed at reducing symptoms, as there is no cure for I-cell disease. Complications of disease include heart failure Heart Failure A heterogeneous condition in which the heart is unable to pump out sufficient blood to meet the metabolic need of the body. Heart failure can be caused by structural defects, functional abnormalities (ventricular dysfunction), or a sudden overload beyond its capacity. Chronic heart failure is more common than acute heart failure which results from sudden insult to cardiac function, such as myocardial infarction. Total Anomalous Pulmonary Venous Return (TAPVR) and infection, with death occurring in the 1st decade of life.
Last updated: Apr 23, 2025
I-cell disease is a rare lysosomal storage disorder that presents in early childhood with severe skeletal abnormalities and failure to thrive Failure to Thrive Failure to thrive (FTT), or faltering growth, describes suboptimal weight gain and growth in children. The majority of cases are due to inadequate caloric intake; however, genetic, infectious, and oncological etiologies are also common. Failure to Thrive. A mutation Mutation Genetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations in the GNPTA gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics causes a deficiency in the enzyme uridine diphosphate Uridine diphosphate A uracil nucleotide containing a pyrophosphate group esterified to C5 of the sugar moiety. Purine and Pyrimidine Metabolism (UDP)-N-acetylglucosamine-1-phosphotransferase.
Clinical features evident by 6 months of age:
Patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship present in 1st year of life with:
Progressive, severe psychomotor impairment leads to death in early childhood.
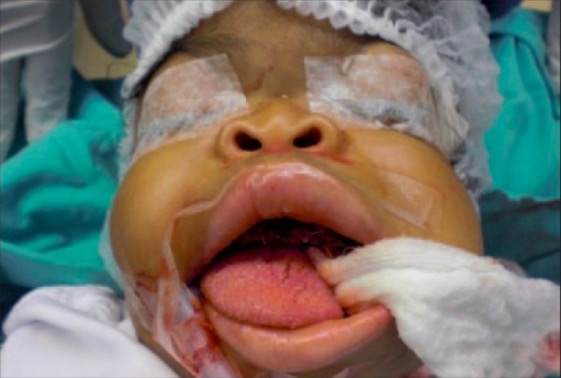
Facial features seen in I-cell disease:
Characteristic coarse facial features are distinctive of patients with mucolipidosis II. These features become more evident with time and can present a challenge for intubation during surgery.
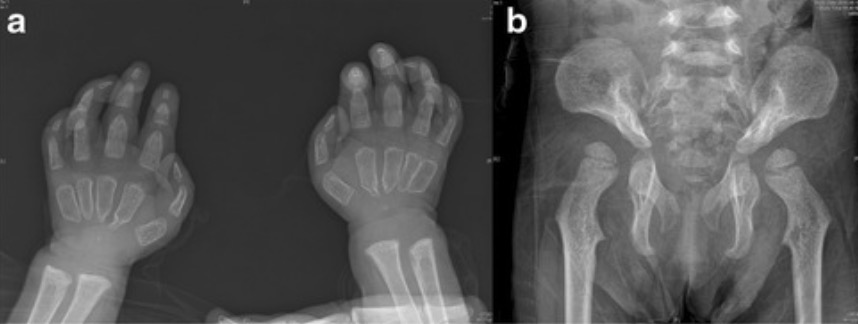
Skeletal abnormalities in patients with I-cell disease:
Patients with mucolipidosis II often present with skeletal abnormalities. Commonly seen defects are restriction of the joints. These X-rays show broad, undermodeled proximal phalanges and proximal pointing of metacarpals (a) and hip dysplasia (b).
Prenatal diagnosis:
Diagnostic tests Diagnostic tests Diagnostic tests are important aspects in making a diagnosis. Some of the most important epidemiological values of diagnostic tests include sensitivity and specificity, false positives and false negatives, positive and negative predictive values, likelihood ratios, and pre-test and post-test probabilities. Epidemiological Values of Diagnostic Tests performed after delivery:
No curative treatment is available for I-cell disease. Management is symptomatic and supportive:
Bone marrow transplantation Bone marrow transplantation Transfer of hematopoietic stem cells from bone marrow or blood between individuals within the same species (homologous transplantation) or transfer within the same individual (autologous transplantation). Hematopoietic stem cell transplantation has been used as an alternative to bone marrow transplantation in the treatment of a variety of neoplasms. Organ Transplantation may be helpful as a remedy for neurologic degeneration (currently under investigation).
Genetic counseling Genetic Counseling An educational process that provides information and advice to individuals or families about a genetic condition that may affect them. The purpose is to help individuals make informed decisions about marriage, reproduction, and other health management issues based on information about the genetic disease, the available diagnostic tests, and management programs. Psychosocial support is usually offered. Myotonic Dystrophies should be offered to families considering having additional children.