La enfermedad de las células de inclusión (enfermedad de las células I, mucolipidosis II o ML II) está causada por un defecto en la uridina difosfato (UDP)-N-acetilgucosamina-1-fosfotransferasa, una enzima que transfiere fosfato a los residuos de manosa en proteínas específicas. Los lisosomas deben tener esta proteína, ya que es responsable de la descomposición de los oligosacáridos, los lípidos y los glicosaminoglicanos. La deficiencia de esta enzima provoca la acumulación de estas sustancias químicas en el cuerpo, causando "inclusiones celulares". Los LOS Neisseria pacientes presentan retraso en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el desarrollo en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el 1er año de vida. Los LOS Neisseria análisis de sangre para la actividad enzimática y la presencia de cuerpos de inclusión son diagnósticos. El tratamiento está dirigido a reducir los LOS Neisseria síntomas, ya que no hay cura para la enfermedad de las células I. Las complicaciones de la enfermedad incluyen insuficiencia cardíaca e infección, ocurriendo la muerte en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la 1ra década de vida.
Last updated: Apr 23, 2025
La enfermedad de las células I es un raro trastorno de almacenamiento lisosómico que se presenta en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la primera infancia con graves anomalías esqueléticas y retraso en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el crecimiento. Una mutación en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el gen GNPTA causa una deficiencia en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la enzima uridina difosfato (UDP)-N-acetilglucosamina-1-fosfotransferasa.
Características clínicas evidentes a los LOS Neisseria 6 meses de edad:
Los LOS Neisseria pacientes se presentan en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el 1er año de vida con:
El deterioro psicomotor progresivo y grave conduce a la muerte en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la infancia temprana.
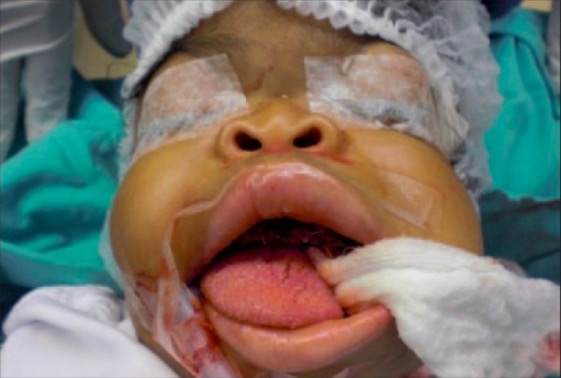
Rasgos faciales observados en la enfermedad de las células I:
Los rasgos faciales gruesos característicos son distintivos de los pacientes con mucolipidosis II. Estas características se hacen más evidentes con el tiempo y pueden suponer un reto para la intubación durante la cirugía.
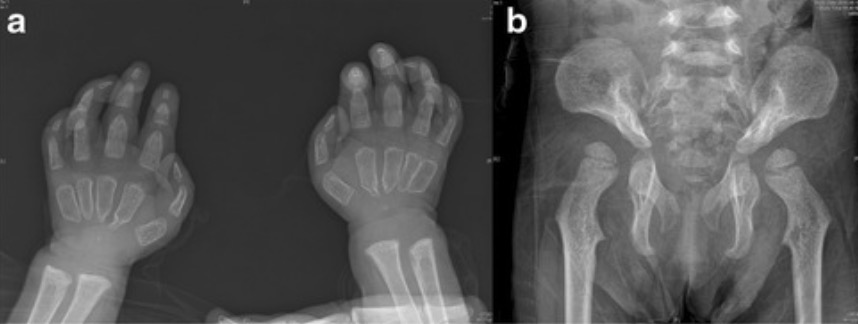
Anomalías esqueléticas en pacientes con enfermedad de células I:
Los pacientes con mucolipidosis II suelen presentar anomalías esqueléticas. Los defectos más comunes son la restricción de las articulaciones. Estas radiografías muestran falanges proximales anchas y poco modeladas y el apuntamiento proximal de los metacarpianos (a) y la displasia de cadera (b).
Diagnóstico prenatal:
Pruebas diagnósticas realizadas después del parto:
No existe ningún tratamiento curativo para la enfermedad de las células I. El tratamiento es sintomático y de soporte:
El trasplante de médula ósea puede ser útil como remedio para la degeneración neurológica (actualmente en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum investigación).
Debe ofrecerse asesoramiento genético a las familias que se plantean tener más hijos.