


A doença das células de inclusão (doença de I-cell, mucolipidose II, ou ML II) é causada por um defeito no difosfato de uridina (UDP)-N-acetilglucosamina-1-fosfotransferase, uma enzima que transfere fosfato para resíduos de manose em proteínas específicas. Os lisossomas devem ter essa proteína, uma vez que ela é responsável pela quebra de oligossacarídeos, lípidos e glucosaminoglicanos. Uma deficiência nesta enzima resulta na acumulação destas substâncias químicas no organismo, causando "células de inclusão". Os pacientes apresentam-se durante o 1.º ano de vida com má progressão estaturo-ponderal e atraso no desenvolvimento. Análises sanguíneas para a atividade enzimática e a presença de organismos de inclusão são diagnósticos. O tratamento visa reduzir os sintomas, já que não há cura para a doença de I-cell. Complicações da doença incluem insuficiência cardíaca e infeção, com a morte a ocorrer na 1.ª década de vida.
Last updated: Apr 23, 2025
A doença de I-cell é uma doença rara no depósito lisossómico que se apresenta na infância com anomalias esqueléticas graves e má progressão estaturo-ponderal. Uma mutação no gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics GNPTA causa uma deficiência na enzima difosfato de uridina (UDP)-N-acetilglucosamina-1-fosfotransferase.
Características clínicas evidentes aos 6 meses de idade:
Pacientes apresentam-se no 1.º ano de vida com:
Uma deficiência psicomotora progressiva e grave leva à morte na infância.
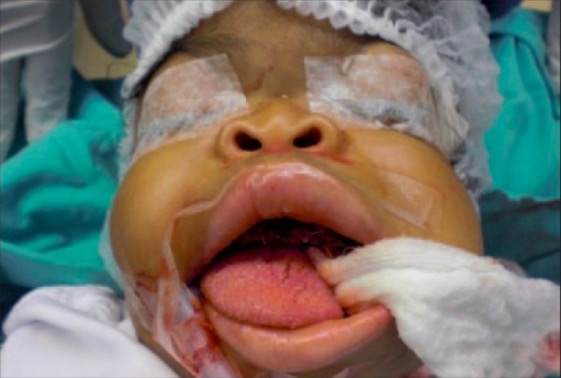
Características faciais vistas na doença de I-cell:
Os elementos faciais rugosos característicos dos pacientes com mucolipidose II. Estas características tornam-se mais evidentes com o tempo e podem representar um desafio para a entubação durante a cirurgia.
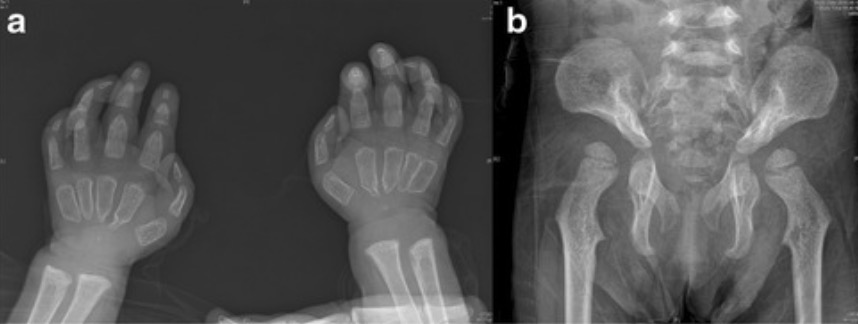
Anomalias esqueléticas em pacientes com doença de I-cell:
Os doentes com mucolipidose II apresentam frequentemente anomalias esqueléticas. Defeitos frequentemente observados incluem restrição das articulações. Estas radiografias mostram falanges proximais largas e não modeladas, e apontamento proximal de metacarpos. (a) e displasia da anca (b).
Diagnóstico pré-natal:
Testes Testes Gonadal Hormones diagnósticos realizados após o parto:
Não existe cura disponível para a doença de I-cell. O tratamento é sintomático e de apoio:
O transplante de medula óssea pode ser útil na solução da degeneração neurológica (atualmente em investigação).
O aconselhamento genético deve ser oferecido às famílias que consideram ter novos filhos.
