Krabbe disease, also known as globoid cell leukodystrophy or galactosylceramide lipidosis, is a rare autosomal recessive Autosomal recessive Autosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal recessive diseases are only expressed when 2 copies of the recessive allele are inherited. Autosomal Recessive and Autosomal Dominant Inheritance lysosomal storage disorder caused by a deficiency of the enzyme galactocerebrosidase. Accumulation of galactocerebroside results in destruction of myelin-producing cells throughout the peripheral and central nervous systems, leading to demyelination Demyelination Multiple Sclerosis and clinical symptoms. The disease is classified into infantile-onset (classic) and late-onset subtypes. Clinical manifestations for classic Krabbe disease include irritability, hypertonia Hypertonia Abnormal increase in skeletal or smooth muscle tone. Skeletal muscle hypertonicity may be associated with pyramidal tract lesions or basal ganglia diseases. Neurological Examination, difficulty feeding, failure to thrive Failure to Thrive Failure to thrive (FTT), or faltering growth, describes suboptimal weight gain and growth in children. The majority of cases are due to inadequate caloric intake; however, genetic, infectious, and oncological etiologies are also common. Failure to Thrive, and rapid neurodegeneration. Death occurs from infection or respiratory failure Respiratory failure Respiratory failure is a syndrome that develops when the respiratory system is unable to maintain oxygenation and/or ventilation. Respiratory failure may be acute or chronic and is classified as hypoxemic, hypercapnic, or a combination of the two. Respiratory Failure. Diagnosis is made by measuring enzyme activity. There is no cure for Krabbe disease. Management is supportive. Stem cell transplantation is an option for some infants.
Last updated: Apr 23, 2025
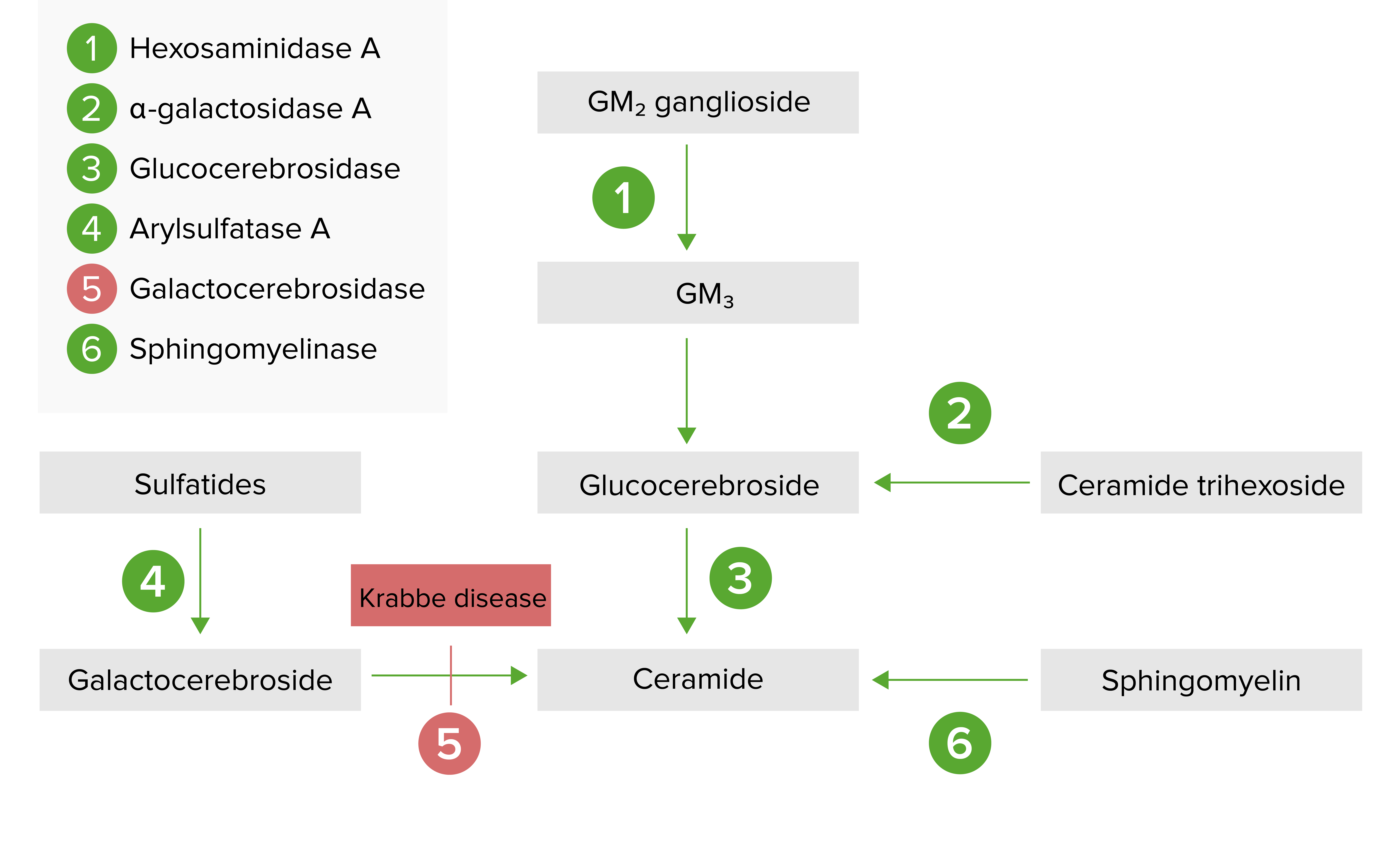
The lysosomal storage pathway:
Krabbe disease results from galactocerebrosidase enzyme deficiency (step 5) and the subsequent buildup of galactocerebroside.
The classic, infantile form progresses through the following stages:
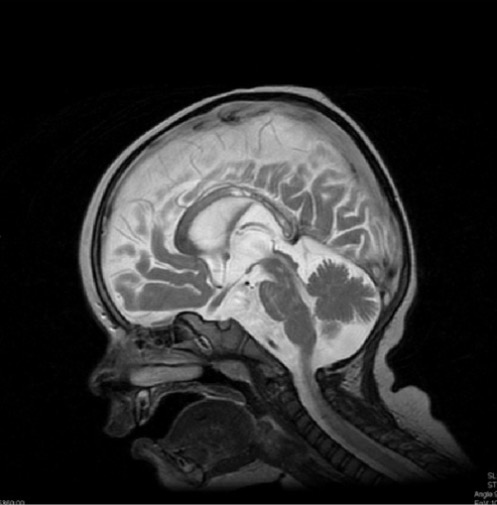
A 13-month-old boy with diffuse demyelination (hyperintensity) of the corpus callosum on MRI, characteristic of Krabbe disease
Image: “Krabbe disease on imaging” by Department of Diagnostic Imaging, Institute of Mother and Child, ul. Kasprzaka, 17a, 01-211 Warsaw, Poland. License: CC BY 3.0