


A doença de Krabbe, também conhecida como leucodistrofia de células globoides ou lipidose de galactosilceramida, é uma doença autossómica recessiva rara de armazenamento lisossómico, causada por uma deficiência da enzima galactocerebrosidase Galactocerebrosidase An enzyme that hydrolyzes galactose from ceramide monohexosides. Deficiency of this enzyme may cause globoid cell leukodystrophy. Krabbe Disease. A acumulação de galactocerebrosídeo resulta na destruição de células produtoras de mielina em todo o sistema nervoso periférico e central, levando à desmielinização e sintomas clínicos. A doença é classificada nos subtipos de início infantil (clássico) e de início tardio. As manifestações clínicas da doença de Krabbe clássica incluem irritabilidade, hipertonia, dificuldade de alimentação, má progressão estaturo-ponderal e neurodegeneração rápida. A morte ocorre por infeção ou insuficiência respiratória. O diagnóstico é feito através da medição da atividade enzimática. Não há cura para a doença de Krabbe. O tratamento é de suporte. O transplante de células estaminais é uma opção para alguns bebés.
Last updated: Apr 23, 2025
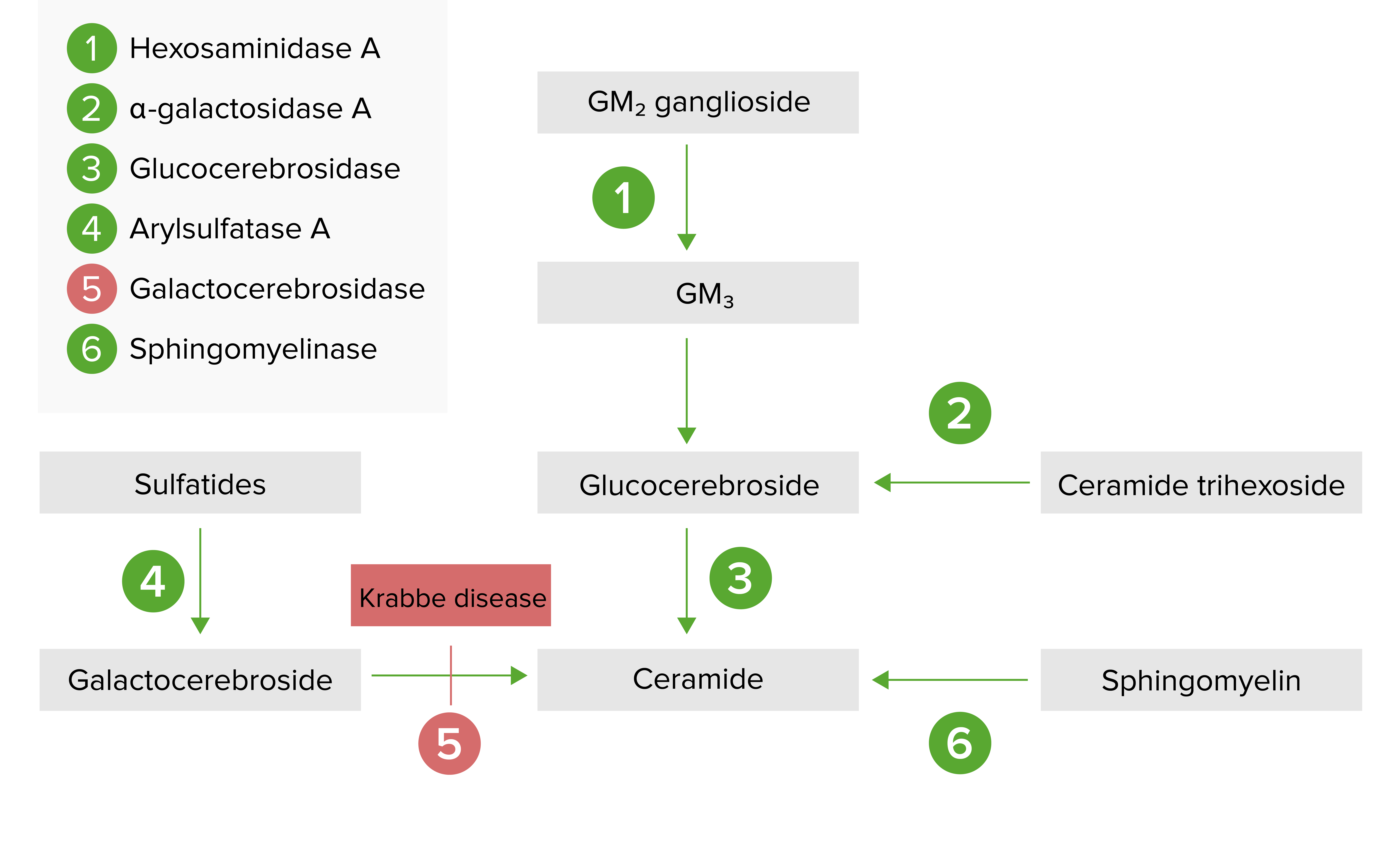
A via de armazenamento lisossomal:
A doença de Krabbe resulta da deficiência da enzima galactocerebrosidase (passo 5) e da subsequente acumulação de galactocerebrosídeo.
A forma clássica, infantil, evolui através das seguintes etapas:
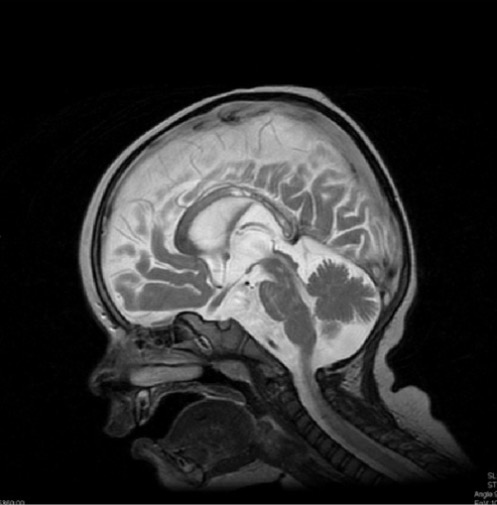
Um menino de 13 meses com desmielinização difusa (hiperintensidade) do corpo caloso na ressonância magnética, característica da doença de Krabbe
Imagem: “Krabbe disease on imaging” por Department of Diagnostic Imaging, Institute of Mother and Child, ul. Kasprzaka, 17a, 01-211 Warsaw, Poland. Licença: CC BY 3.0