La enfermedad de Krabbe, también conocida como leucodistrofia de células globoides o lipidosis de galactosilceramida, es un raro trastorno autosómico recesivo que afecta el almacenamiento lisosómico, causado por una deficiencia de la enzima galactocerebrosidasa. La acumulación de galactocerebrósido provoca la destrucción de las células productoras de mielina en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum todo el sistema nervioso periférico y central, lo que conduce a la desmielinización y a los LOS Neisseria síntomas clínicos. La enfermedad se clasifica en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum subtipos de inicio infantil (clásico) y de inicio tardío. Las manifestaciones clínicas de la enfermedad de Krabbe clásica incluyen: irritabilidad, hipertonía, dificultad para alimentarse, retraso en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el desarrollo y rápida neurodegeneración. La muerte se produce por una infección, o por insuficiencia respiratoria. El diagnóstico se realiza midiendo la actividad enzimática. No existe cura para la leucodistrofia de células globoides. El tratamiento es de soporte. El trasplante de células madre es una opción para algunos lactantes.
Last updated: Apr 23, 2025
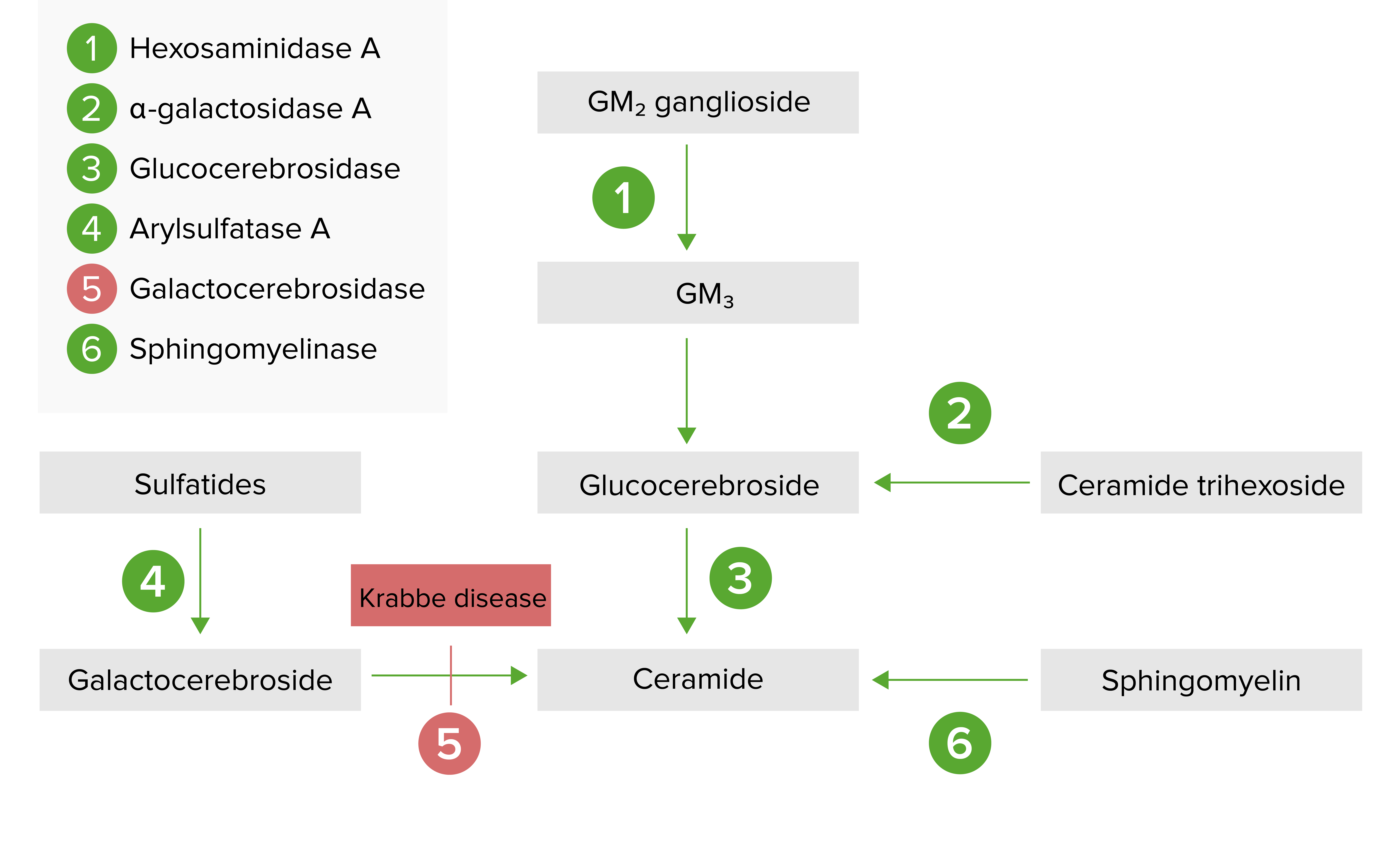
La vía de almacenamiento lisosómico:
La enfermedad de Krabbe, es el resultado de la deficiencia de la enzima galactocerebrosidasa (paso 5) y la subsiguiente acumulación de galactocerebrósido.
La forma clásica, infantil, progresa a través de las siguientes etapas:
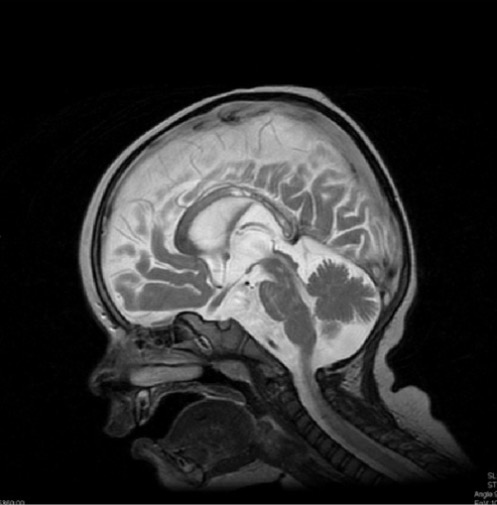
Un niño de 13 meses con desmielinización difusa (hiperintensidad) del cuerpo calloso en la RM, característica de la enfermedad de Krabbe.
Imagen: “Krabbe disease on imaging” por Department of Diagnostic Imaging, Institute of Mother and Child, ul. Kasprzaka, 17a, 01-211 Warsaw, Poland. Licencia: CC BY 3.0