Rett syndrome is a rare genetic neurological and developmental disorder that affects the development of the brain. There are various stages of Rett syndrome characterized by different symptoms and clinical signs. These stages include increasing problems with the use of muscles that control movement, coordination, and communication. Trofinetide is the first disease-modifying treatment for Rett syndrome (approved in 2023).
Rett syndrome is a rare genetic disorder that is inherited in an X-linkedX-linkedGenetic diseases that are linked to gene mutations on the X chromosome in humans or the X chromosome in other species. Included here are animal models of human X-linked diseases.Common Variable Immunodeficiency (CVID) dominant fashion.
Second most common cause of severe intellectual disabilityDisabilityDetermination of the degree of a physical, mental, or emotional handicap. The diagnosis is applied to legal qualification for benefits and income under disability insurance and to eligibility for social security and workman’s compensation benefits.ABCDE Assessment after Down syndromeDown syndromeDown syndrome, or trisomy 21, is the most common chromosomal aberration and the most frequent genetic cause of developmental delay. Both boys and girls are affected and have characteristic craniofacial and musculoskeletal features, as well as multiple medical anomalies involving the cardiac, gastrointestinal, ocular, and auditory systems.Down syndrome (Trisomy 21) (trisomy 21Trisomy 21Down syndrome, or trisomy 21, is the most common chromosomal aberration and the most frequent genetic cause of developmental delay. Both boys and girls are affected and have characteristic craniofacial and musculoskeletal features, as well as multiple medical anomalies involving the cardiac, gastrointestinal, ocular, and auditory systems.Down syndrome (Trisomy 21))
90%–95% of cases are caused by mutations of the MECP2 geneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics (methyl-CpG binding protein 2).
MutationMutationGenetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations affects protein production critical for brainBrainThe part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem.Nervous System: Anatomy, Structure, and Classification development
The mutated copy is often associated with the paternal alleleAlleleVariant forms of the same gene, occupying the same locus on homologous chromosomes, and governing the variants in production of the same gene product.Basic Terms of Genetics since it occurs during spermatogenesisSpermatogenesisThe process of germ cell development in the male from the primordial germ cells, through spermatogonia; spermatocytes; spermatids; to the mature haploid spermatozoa.Gametogenesis (only females inherit the paternal X chromosomeX chromosomeThe female sex chromosome, being the differential sex chromosome carried by half the male gametes and all female gametes in human and other male-heterogametic species.Basic Terms of Genetics, as the paternal Y chromosomeY chromosomeThe male sex chromosome, being the differential sex chromosome carried by half the male gametes and none of the female gametes in humans and in some other male-heterogametic species in which the homologue of the X chromosome has been retained.Basic Terms of Genetics determines the male sexSexThe totality of characteristics of reproductive structure, functions, phenotype, and genotype, differentiating the male from the female organism.Gender Dysphoria of the fetus).
Females are almost exclusively affected.
Most males with this mutationMutationGenetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations are believed to die in utero or during early infancy.
IncidenceIncidenceThe number of new cases of a given disease during a given period in a specified population. It also is used for the rate at which new events occur in a defined population. It is differentiated from prevalence, which refers to all cases in the population at a given time.Measures of Disease Frequency is 1 in 10,000 girls by age 12 in the United States.
Some boys may have a different mutationMutationGenetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations, resulting in a less destructive form of Rett syndrome.
Life expectancyLife expectancyBased on known statistical data, the number of years which any person of a given age may reasonably expected to live.Population Pyramids is higher.
Lower risk for intellectual and developmental problems
Clinical Presentation and Diagnosis
Clinical presentation
Individuals with Rett syndrome often have normal development until 7–24 months of age.
Developmental stagnation occurs between 6 and 18 months of age.
BrainBrainThe part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem.Nervous System: Anatomy, Structure, and Classification growth slows after birth.
MicrocephalyMicrocephalyA congenital abnormality in which the cerebrum is underdeveloped, the fontanels close prematurely, and, as a result, the head is small. (desk reference for neuroscience, 2nd ed. ).Fetal Alcohol Spectrum Disorder
Reduced handHandThe hand constitutes the distal part of the upper limb and provides the fine, precise movements needed in activities of daily living. It consists of 5 metacarpal bones and 14 phalanges, as well as numerous muscles innervated by the median and ulnar nerves. Hand: Anatomy control
Repetitive, purposeless handHandThe hand constitutes the distal part of the upper limb and provides the fine, precise movements needed in activities of daily living. It consists of 5 metacarpal bones and 14 phalanges, as well as numerous muscles innervated by the median and ulnar nerves. Hand: Anatomy movements (hand-wringing, squeezing, clapping, tapping, rubbing)
Unusual eye movements
Intense staring
Excessive blinking
Crossed eyes or closing one eye at a time
Gait ataxiaGait ataxiaImpairment of the ability to coordinate the movements required for normal ambulation (walking) which may result from impairments of motor function or sensory feedback. This condition may be associated with brain diseases (including cerebellar diseases and basal ganglia diseases); spinal cord diseases; or peripheral nervous system diseases.Friedreich Ataxia
DystoniaDystoniaDystonia is a hyperkinetic movement disorder characterized by the involuntary contraction of muscles, resulting in abnormal postures or twisting and repetitive movements. Dystonia can present in various ways as may affect many different skeletal muscle groups. Dystonia (sustained muscle contractions associated with uncontrolled movements and postures)
Breathing problems
Breath-holding
Abnormally rapid breathing (hyperventilationHyperventilationA pulmonary ventilation rate faster than is metabolically necessary for the exchange of gases. It is the result of an increased frequency of breathing, an increased tidal volume, or a combination of both. It causes an excess intake of oxygen and the blowing off of carbon dioxide.Respiratory Alkalosis)
Forceful exhalation of air or salivaSalivaThe clear, viscous fluid secreted by the salivary glands and mucous glands of the mouth. It contains mucins, water, organic salts, and ptyalin.Salivary Glands: Anatomy
SwallowingSwallowingThe act of taking solids and liquids into the gastrointestinal tract through the mouth and throat.Gastrointestinal Motility air (aerophagia)
Abnormal behaviors
Infants are “placid,” demanding little to no attentionAttentionFocusing on certain aspects of current experience to the exclusion of others. It is the act of heeding or taking notice or concentrating.Psychiatric Assessment from parents
Sudden, odd facial expressions
Long bouts of laughter
HandHandThe hand constitutes the distal part of the upper limb and provides the fine, precise movements needed in activities of daily living. It consists of 5 metacarpal bones and 14 phalanges, as well as numerous muscles innervated by the median and ulnar nerves. Hand: Anatomy licking
Grasping of hair or clothing
Increasingly agitate, irritable, and restless with age
ApraxiaApraxiaA group of cognitive disorders characterized by the inability to perform previously learned skills that cannot be attributed to deficits of motor or sensory function. The two major subtypes of this condition are ideomotor and ideational apraxia, which refers to loss of the ability to mentally formulate the processes involved with performing an action. For example, dressing apraxia may result from an inability to mentally formulate the act of placing clothes on the body. Apraxias are generally associated with lesions of the dominant parietal lobe and supramarginal gyrus.Cranial Nerve Palsies (inability to perform learned movements on command)
TeethTeethNormally, an adult has 32 teeth: 16 maxillary and 16 mandibular. These teeth are divided into 4 quadrants with 8 teeth each. Each quadrant consists of 2 incisors (dentes incisivi), 1 canine (dens caninus), 2 premolars (dentes premolares), and 3 molars (dentes molares). Teeth are composed of enamel, dentin, and dental cement.Teeth: Anatomy grinding
Abnormal sleepSleepA readily reversible suspension of sensorimotor interaction with the environment, usually associated with recumbency and immobility.Physiology of Sleep patterns
Irregular heartbeat (can result in sudden death)
ScoliosisScoliosisScoliosis is a structural alteration of the vertebral column characterized by a lateral spinal curvature of greater than 10 degrees in the coronal plane. Scoliosis can be classified as idiopathic (in most cases) or secondary to underlying conditions. Scoliosis
Tremors and seizuresSeizuresA seizure is abnormal electrical activity of the neurons in the cerebral cortex that can manifest in numerous ways depending on the region of the brain affected. Seizures consist of a sudden imbalance that occurs between the excitatory and inhibitory signals in cortical neurons, creating a net excitation. The 2 major classes of seizures are focal and generalized. Seizures (with abnormal EEGEEGSeizures tracing)
Diagnosis
Diagnosis is based on clinical presentation, a detailed patient history, and thorough clinical evaluation.
Genetic testingGenetic TestingDetection of a mutation; genotype; karyotype; or specific alleles associated with genetic traits, heritable diseases, or predisposition to a disease, or that may lead to the disease in descendants. It includes prenatal genetic testing.Myotonic Dystrophies for MECP2geneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics mutations may be used to confirm and/or rule out similar syndromes.
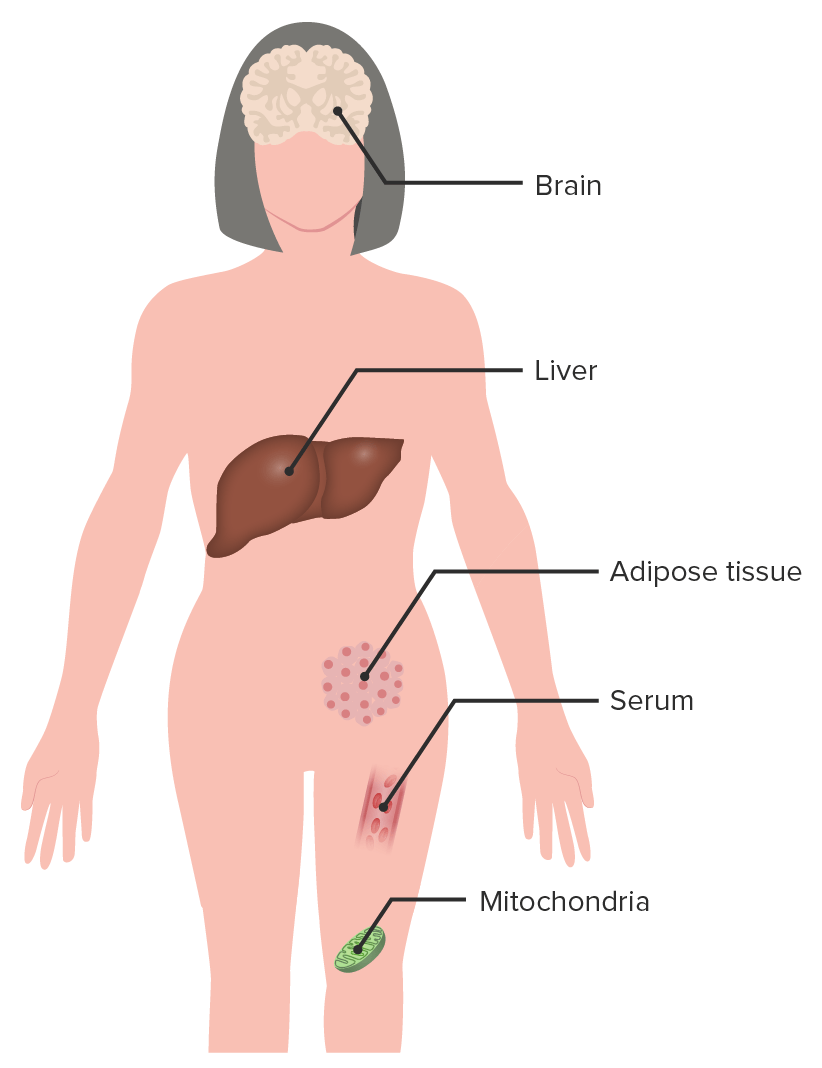
Image showing the various systems affected in Rett Syndrome: Multiple systems are affected and individuals display a varied clinical picture.
Image by Lecturio.
Clinical features/signs in RETT syndrome
BrainBrainThe part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem.Nervous System: Anatomy, Structure, and Classification:
Abnormalities in neurometabolites
Altered carbohydrate metabolism in CSF
Increased pyruvatePyruvateDerivatives of pyruvic acid, including its salts and esters.Glycolysis and lactate in CSF
Elevated lipidsLipidsLipids are a diverse group of hydrophobic organic molecules, which include fats, oils, sterols, and waxes.Fatty Acids and Lipids
Reduced choline phospholipid turnover
Oxidative changes
LiverLiverThe liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy:
InflammationInflammationInflammation is a complex set of responses to infection and injury involving leukocytes as the principal cellular mediators in the body’s defense against pathogenic organisms. Inflammation is also seen as a response to tissue injury in the process of wound healing. The 5 cardinal signs of inflammation are pain, heat, redness, swelling, and loss of function. Inflammation of the gallbladderGallbladderThe gallbladder is a pear-shaped sac, located directly beneath the liver, that sits on top of the superior part of the duodenum. The primary functions of the gallbladder include concentrating and storing up to 50 mL of bile. Gallbladder and Biliary Tract: Anatomy
Elevated triglyceridesTriglyceridesFatty Acids and Lipids (fatty liverLiverThe liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy)
Adipose tissueAdipose tissueAdipose tissue is a specialized type of connective tissue that has both structural and highly complex metabolic functions, including energy storage, glucose homeostasis, and a multitude of endocrine capabilities. There are three types of adipose tissue, white adipose tissue, brown adipose tissue, and beige or “brite” adipose tissue, which is a transitional form.Adipose Tissue: Histology:
Abnormal glucoseGlucoseA primary source of energy for living organisms. It is naturally occurring and is found in fruits and other parts of plants in its free state. It is used therapeutically in fluid and nutrient replacement.Lactose IntolerancetoleranceTolerancePharmacokinetics and Pharmacodynamics test
Insulin resistanceInsulin resistanceDiminished effectiveness of insulin in lowering blood sugar levels: requiring the use of 200 units or more of insulin per day to prevent hyperglycemia or ketosis.Diabetes Mellitus
Increased leptinLeptinA 16-kda peptide hormone secreted from white adipocytes. Leptin serves as a feedback signal from fat cells to the central nervous system in regulation of food intake, energy balance, and fat storage.Adipose Tissue: Histology, pyruvatePyruvateDerivatives of pyruvic acid, including its salts and esters.Glycolysis, and lactate
Decreased ghrelinGhrelinA 28-amino acid, Acylated, orexigenic peptide that is a ligand for growth hormone secretagogue receptors. Ghrelin is widely expressed but primarily in the stomach in the adults. Ghrelin acts centrally to stimulate growth hormone secretion and food intake, and peripherally to regulate energy homeostasis. Its large precursor protein, known as appetite-regulating hormone or motilin-related peptide, contains ghrelin and obestatin.Glucagonoma
Increase ammoniaAmmoniaA colorless alkaline gas. It is formed in the body during decomposition of organic materials during a large number of metabolically important reactions. Note that the aqueous form of ammonia is referred to as ammonium hydroxide.Acid-Base Balance
MitochondriaMitochondriaSemiautonomous, self-reproducing organelles that occur in the cytoplasm of all cells of most, but not all, eukaryotes. Each mitochondrion is surrounded by a double limiting membrane. The inner membrane is highly invaginated, and its projections are called cristae. Mitochondria are the sites of the reactions of oxidative phosphorylation, which result in the formation of ATP. They contain distinctive ribosomes, transfer RNAs; amino Acyl tRNA synthetases; and elongation and termination factors. Mitochondria depend upon genes within the nucleus of the cells in which they reside for many essential messenger RNAs. Mitochondria are believed to have arisen from aerobic bacteria that established a symbiotic relationship with primitive protoeukaryotes.The Cell: Organelles:
Abnormal mitochondrial structure
Altered function of the electron transport chainElectron transport chainThe electron transport chain (ETC) sends electrons through a series of proteins, which generate an electrochemical proton gradient that produces energy in the form of adenosine triphosphate (ATP).Electron Transport Chain (ETC)
Increase in oxidative stressOxidative stressA disturbance in the prooxidant-antioxidant balance in favor of the former, leading to potential damage. Indicators of oxidative stress include damaged DNA bases, protein oxidation products, and lipid peroxidation products.Cell Injury and Death
Stages
Stage I: Developmental arrest (6–18 months of age)
Signs and symptoms are subtle.
Lasts for a few months up to a year
May present with less eye contact and placidity
The achievement of developmental milestonesDevelopmental milestonesDevelopmental milestones are the skills or abilities that most children are able to perform when they reach a certain age. Understanding the appropriate milestones and at what age they are reached helps clinicians identify symptoms of delayed development. Developmental milestones are divided into 5 important domains: gross motor, fine motor, language, social, and cognitive. Developmental Milestones and Normal Growth becomes slowly and increasingly delayed.
Children lose the ability to perform previously acquired skills.
Loss can be rapid or more gradual.
Reduced head growth
Abnormal handHandThe hand constitutes the distal part of the upper limb and provides the fine, precise movements needed in activities of daily living. It consists of 5 metacarpal bones and 14 phalanges, as well as numerous muscles innervated by the median and ulnar nerves. Hand: Anatomy movements (“wringing”)
SeizuresSeizuresA seizure is abnormal electrical activity of the neurons in the cerebral cortex that can manifest in numerous ways depending on the region of the brain affected. Seizures consist of a sudden imbalance that occurs between the excitatory and inhibitory signals in cortical neurons, creating a net excitation. The 2 major classes of seizures are focal and generalized. Seizures may begin in this stage.
Stage IV: Late motorMotorNeurons which send impulses peripherally to activate muscles or secretory cells.Nervous System: Histology deterioration (> 10 years)
Marked by reduced mobility
Muscle weakness
Joint contracturesContracturesProlonged shortening of the muscle or other soft tissue around a joint, preventing movement of the joint.Wound Healing
ScoliosisScoliosisScoliosis is a structural alteration of the vertebral column characterized by a lateral spinal curvature of greater than 10 degrees in the coronal plane. Scoliosis can be classified as idiopathic (in most cases) or secondary to underlying conditions. Scoliosis
Complications
SleepSleepA readily reversible suspension of sensorimotor interaction with the environment, usually associated with recumbency and immobility.Physiology of Sleep problems cause significant fatigueFatigueThe state of weariness following a period of exertion, mental or physical, characterized by a decreased capacity for work and reduced efficiency to respond to stimuli.Fibromyalgia and restlessness.
Difficulty eating leads to malnutritionMalnutritionMalnutrition is a clinical state caused by an imbalance or deficiency of calories and/or micronutrients and macronutrients. The 2 main manifestations of acute severe malnutrition are marasmus (total caloric insufficiency) and kwashiorkor (protein malnutrition with characteristic edema).Malnutrition in children in resource-limited countries.
Bowel and bladderBladderA musculomembranous sac along the urinary tract. Urine flows from the kidneys into the bladder via the ureters, and is held there until urination.Pyelonephritis and Perinephric Abscess problems
ConstipationConstipationConstipation is common and may be due to a variety of causes. Constipation is generally defined as bowel movement frequency < 3 times per week. Patients who are constipated often strain to pass hard stools. The condition is classified as primary (also known as idiopathic or functional constipation) or secondary, and as acute or chronic. Constipation
Gastroesophageal reflux diseaseGastroesophageal Reflux DiseaseGastroesophageal reflux disease (GERD) occurs when the stomach acid frequently flows back into the esophagus. This backwash (acid reflux) can irritate the lining of the esophagus, causing symptoms such as retrosternal burning pain (heartburn). Gastroesophageal Reflux Disease (GERD) (GERDGERDGastroesophageal reflux disease (GERD) occurs when the stomach acid frequently flows back into the esophagus. This backwash (acid reflux) can irritate the lining of the esophagus, causing symptoms such as retrosternal burning pain (heartburn). Gastroesophageal Reflux Disease (GERD))
Bowel or urinary incontinenceUrinary incontinenceUrinary incontinence (UI) is involuntary loss of bladder control or unintentional voiding, which represents a hygienic or social problem to the patient. Urinary incontinence is a symptom, a sign, and a disorder. The 5 types of UI include stress, urge, mixed, overflow, and functional.Urinary Incontinence
GallbladderGallbladderThe gallbladder is a pear-shaped sac, located directly beneath the liver, that sits on top of the superior part of the duodenum. The primary functions of the gallbladder include concentrating and storing up to 50 mL of bile. Gallbladder and Biliary Tract: Anatomy disease
Muscle, boneBoneBone is a compact type of hardened connective tissue composed of bone cells, membranes, an extracellular mineralized matrix, and central bone marrow. The 2 primary types of bone are compact and spongy. Bones: Structure and Types, and joint problems
AnxietyAnxietyFeelings or emotions of dread, apprehension, and impending disaster but not disabling as with anxiety disorders.Generalized Anxiety Disorder and problematic behavior that may hinder social functioning
Needing lifelong care and assistance with activities of daily life
Shortened life span (median survival is approximately 45 years of age)
Management
Trofinetide:
1st disease-modifying treatment available
Mechanism not completely understood
Studies demonstrate short-term improvement; long-term improvement not yet established.
ScreeningScreeningPreoperative Care and management of complications (e.g., GERDGERDGastroesophageal reflux disease (GERD) occurs when the stomach acid frequently flows back into the esophagus. This backwash (acid reflux) can irritate the lining of the esophagus, causing symptoms such as retrosternal burning pain (heartburn). Gastroesophageal Reflux Disease (GERD), boneBoneBone is a compact type of hardened connective tissue composed of bone cells, membranes, an extracellular mineralized matrix, and central bone marrow. The 2 primary types of bone are compact and spongy. Bones: Structure and Types density, seizuresSeizuresA seizure is abnormal electrical activity of the neurons in the cerebral cortex that can manifest in numerous ways depending on the region of the brain affected. Seizures consist of a sudden imbalance that occurs between the excitatory and inhibitory signals in cortical neurons, creating a net excitation. The 2 major classes of seizures are focal and generalized. Seizures)
The following conditions are differential diagnoses for Rett syndrome:
Angelman syndromeAngelman syndromeAngelman syndrome (AS) is a rare autosomal neurodevelopmental genetic disorders mapped to a specific region of chromosome 15 attributed to genomic imprinting. A maternally derived chromosome 15 with this deletion results in 15q11-13 maternal deletion syndrome, or AS. Prader-Willi Syndrome and Angelman Syndrome: an autosomal neuro-developmental disease resulting from epigenetic sex-specific genomic imprintingImprintingThe variable phenotypic expression of a gene depending on whether it is of paternal or maternal origin, which is a function of the DNA methylation pattern. Imprinted regions are observed to be more methylated and less transcriptionally active.Epigenetic Regulation and uniparental disomy of paternal chromosome 15Chromosome 15Marfan Syndrome with a simultaneous functional loss of the maternal part. Characterized by inappropriate laughter, seemingly happy demeanor, short attentionAttentionFocusing on certain aspects of current experience to the exclusion of others. It is the act of heeding or taking notice or concentrating.Psychiatric Assessment span, lack of proper speech, use of non-verbal communicationCommunicationThe exchange or transmission of ideas, attitudes, or beliefs between individuals or groups.Decision-making Capacity and Legal Competence skills, ataxiaAtaxiaImpairment of the ability to perform smoothly coordinated voluntary movements. This condition may affect the limbs, trunk, eyes, pharynx, larynx, and other structures. Ataxia may result from impaired sensory or motor function. Sensory ataxia may result from posterior column injury or peripheral nerve diseases. Motor ataxia may be associated with cerebellar diseases; cerebral cortex diseases; thalamic diseases; basal ganglia diseases; injury to the red nucleus; and other conditions.Ataxia-telangiectasia, fine tremors, jerky movements, and severe developmental delay.
Prader-Willi syndromePrader-Willi syndromePrader-Willi syndrome (PWS) is a rare autosomal neurodevelopmental genetic disorders mapped to a specific region of chromosome 15 attributed to genomic imprinting. A paternally derived chromosome 15 with this deletion results in 15q11-13 paternal deletion syndrome, or PWS.Prader-Willi Syndrome and Angelman Syndrome: a rare autosomal neuro-developmental disease resulting from genomic imprintingImprintingThe variable phenotypic expression of a gene depending on whether it is of paternal or maternal origin, which is a function of the DNA methylation pattern. Imprinted regions are observed to be more methylated and less transcriptionally active.Epigenetic Regulation and uniparental disomy of maternal chromosome 15Chromosome 15Marfan Syndrome with a simultaneous functional loss of the parental part. Characterized by hypotoniaHypotoniaDuchenne Muscular Dystrophy, lethargyLethargyA general state of sluggishness, listless, or uninterested, with being tired, and having difficulty concentrating and doing simple tasks. It may be related to depression or drug addiction.Hyponatremia, feeding difficulties, breathing difficulties, and hypogonadismHypogonadismHypogonadism is a condition characterized by reduced or no sex hormone production by the testes or ovaries. Hypogonadism can result from primary (hypergonadotropic) or secondary (hypogonadotropic) failure. Symptoms include infertility, increased risk of osteoporosis, erectile dysfunction, decreased libido, and regression (or absence) of secondary sexual characteristics.Hypogonadism at birth; with scoliosisScoliosisScoliosis is a structural alteration of the vertebral column characterized by a lateral spinal curvature of greater than 10 degrees in the coronal plane. Scoliosis can be classified as idiopathic (in most cases) or secondary to underlying conditions. Scoliosis, strabismusStrabismusStrabismus is the misalignment of the eyes while fixating the gaze on an object. Strabismus can be idiopathic, but it may also be caused by cerebral palsy, uncorrected refractive errors, and extraocular muscle or cranial nerve dysfunction. Strabismus, speech delay, intellectual delays, and behavioral issues during childhood.
Landau-Kleffner syndrome: a rare neurodevelopmental disorder characterized by the loss of language comprehension, aphasiaAphasiaA cognitive disorder marked by an impaired ability to comprehend or express language in its written or spoken form. This condition is caused by diseases which affect the language areas of the dominant hemisphere. Clinical features are used to classify the various subtypes of this condition. General categories include receptive, expressive, and mixed forms of aphasia.Ischemic Stroke, and seizuresSeizuresA seizure is abnormal electrical activity of the neurons in the cerebral cortex that can manifest in numerous ways depending on the region of the brain affected. Seizures consist of a sudden imbalance that occurs between the excitatory and inhibitory signals in cortical neurons, creating a net excitation. The 2 major classes of seizures are focal and generalized. Seizures, with severely abnormal electroencephalogram (EEGEEGSeizures) findings during sleepSleepA readily reversible suspension of sensorimotor interaction with the environment, usually associated with recumbency and immobility.Physiology of Sleep and seizuresSeizuresA seizure is abnormal electrical activity of the neurons in the cerebral cortex that can manifest in numerous ways depending on the region of the brain affected. Seizures consist of a sudden imbalance that occurs between the excitatory and inhibitory signals in cortical neurons, creating a net excitation. The 2 major classes of seizures are focal and generalized. Seizures.
Autism spectrum disorderAutism spectrum disorderAutism spectrum disorder (ASD) is a neurodevelopmental disorder marked by poor social skills, restricted interests/social interactions, and repetitive/stereotyped behaviors. The condition is termed a “spectrum” because of the wide variability in the severity of symptoms exhibited. Autism Spectrum Disorder: a neurodevelopmental disorder with early-childhood onset, marked by poor social skills, restricted social interaction and communicationCommunicationThe exchange or transmission of ideas, attitudes, or beliefs between individuals or groups.Decision-making Capacity and Legal Competence, and repetitive and stereotyped behaviors. Because it is a spectrum, some patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship exhibit impairment in language and intellectual levels, while others may have normal or even advanced levels.
Cerebral palsyPalsyparalysis of an area of the body, thus incapable of voluntary movementCranial Nerve Palsies: a syndrome of motorMotorNeurons which send impulses peripherally to activate muscles or secretory cells.Nervous System: Histology impairment caused by a nonprogressive central nervous systemCentral nervous systemThe main information-processing organs of the nervous system, consisting of the brain, spinal cord, and meninges.Nervous System: Anatomy, Structure, and Classification injury. Classified according to muscle toneMuscle toneThe state of activity or tension of a muscle beyond that related to its physical properties, that is, its active resistance to stretch. In skeletal muscle, tonus is dependent upon efferent innervation.Skeletal Muscle Contraction, distribution, and the presumed time of injury. May also present with sensorySensoryNeurons which conduct nerve impulses to the central nervous system.Nervous System: Histology, cognitive, communicationCommunicationThe exchange or transmission of ideas, attitudes, or beliefs between individuals or groups.Decision-making Capacity and Legal Competence, and behavioral disturbances; epilepsyEpilepsyEpilepsy is a chronic brain disorder marked by recurrent and unprovoked seizures. These seizures can be classified as focal or generalized and idiopathic or secondary to another condition. Clinical presentation correlates to the classification of the epileptic disorder. Epilepsy; and musculoskeletal impairment.
CDKL5 (cyclin-dependent kinase-like 5) deficiency disorder: a rare genetic disorder that is inherited in an X-linkedX-linkedGenetic diseases that are linked to gene mutations on the X chromosome in humans or the X chromosome in other species. Included here are animal models of human X-linked diseases.Common Variable Immunodeficiency (CVID) pattern and characterized by early-onset seizuresSeizuresA seizure is abnormal electrical activity of the neurons in the cerebral cortex that can manifest in numerous ways depending on the region of the brain affected. Seizures consist of a sudden imbalance that occurs between the excitatory and inhibitory signals in cortical neurons, creating a net excitation. The 2 major classes of seizures are focal and generalized. Seizures and severe neurodevelopmental impairment. May also present with scoliosisScoliosisScoliosis is a structural alteration of the vertebral column characterized by a lateral spinal curvature of greater than 10 degrees in the coronal plane. Scoliosis can be classified as idiopathic (in most cases) or secondary to underlying conditions. Scoliosis, visual impairment, sensorySensoryNeurons which conduct nerve impulses to the central nervous system.Nervous System: Histology issues, and various gastrointestinal difficulties.
FOXG1-related disorders or FOXG1 syndrome: a group of disorders caused by a mutationMutationGenetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations of the FOXG1geneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics. Affected individuals develop normally in the perinatal period. This is followed by progressive microcephalyMicrocephalyA congenital abnormality in which the cerebrum is underdeveloped, the fontanels close prematurely, and, as a result, the head is small. (desk reference for neuroscience, 2nd ed. ).Fetal Alcohol Spectrum Disorder, seizuresSeizuresA seizure is abnormal electrical activity of the neurons in the cerebral cortex that can manifest in numerous ways depending on the region of the brain affected. Seizures consist of a sudden imbalance that occurs between the excitatory and inhibitory signals in cortical neurons, creating a net excitation. The 2 major classes of seizures are focal and generalized. Seizures, developmental delays, and severe intellectual disabilityDisabilityDetermination of the degree of a physical, mental, or emotional handicap. The diagnosis is applied to legal qualification for benefits and income under disability insurance and to eligibility for social security and workman’s compensation benefits.ABCDE Assessment. May also present with constipationConstipationConstipation is common and may be due to a variety of causes. Constipation is generally defined as bowel movement frequency < 3 times per week. Patients who are constipated often strain to pass hard stools. The condition is classified as primary (also known as idiopathic or functional constipation) or secondary, and as acute or chronic. Constipation, gastroesophageal reflux, scoliosisScoliosisScoliosis is a structural alteration of the vertebral column characterized by a lateral spinal curvature of greater than 10 degrees in the coronal plane. Scoliosis can be classified as idiopathic (in most cases) or secondary to underlying conditions. Scoliosis, footFootThe foot is the terminal portion of the lower limb, whose primary function is to bear weight and facilitate locomotion. The foot comprises 26 bones, including the tarsal bones, metatarsal bones, and phalanges. The bones of the foot form longitudinal and transverse arches and are supported by various muscles, ligaments, and tendons.Foot: Anatomy deformities, and stereotypic handHandThe hand constitutes the distal part of the upper limb and provides the fine, precise movements needed in activities of daily living. It consists of 5 metacarpal bones and 14 phalanges, as well as numerous muscles innervated by the median and ulnar nerves. Hand: Anatomy movements.
References
Porter, R. S., Kaplan, J. L., Lynn, R. B., & Reddy, M. T. (Eds.). (2018). The Merck manual of diagnosis and therapy (20th ed.). Merck Sharp & Dohme Corp.
Neul, J. L., & Kaufmann, W. E. (2023). Rett syndrome: Clinical diagnosis and management. Nature Medicine, 29(5), 953-960. https://doi.org/10.1038/s41591-023-02398-1
Pini, G., & Giannini, C. (2022). Genetics of Rett Syndrome: Mechanisms and Implications (2nd ed.). Springer.
Sellars, E. A., & Hopkin, R. J. (2021). Pediatric Genetics (3rd ed.). Springer.
Zeldin, R. K., & Schwartz, M. (2022). Immunology Made Ridiculously Simple. MedMaster Inc.
Halbach, N. S. J., Smeets, E. E. J., van den Braak, N., et al. (2012). Genotype-phenotype relationships as prognosticators in Rett syndrome should be handled with care in clinical practice. American Journal of Medical Genetics – Part A, 158(2), 340–350.https://doi.org/10.1002/ajmg.a.34418