El síndrome de Rett es un raro trastorno genético neurológico y del desarrollo que afecta alALAmyloidosis desarrollo del cerebro. Existen varias etapas del síndrome de Rett que se caracterizan por diferentes síntomas y signos clínicos. Estas etapas incluyen problemas crecientes con el uso de losLOSNeisseria músculos que controlan el movimiento, la coordinación y la comunicación. Trofinetide es el primer tratamiento modificador de la enfermedad para el síndrome de Rett (aprobado enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum 2023).
El síndrome de Rett es un raro trastorno genético que se hereda de forma dominante ligada alALAmyloidosis cromosoma X.
Es la segunda causa más frecuente de retraso enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el desarrollo grave después del síndrome de Down (trisomía 21)
Las mutaciones genéticas que causan la enfermedad se producen de forma esporádica (99% son de novo)
El 90%–95% de losLOSNeisseria casos están causados por mutaciones del gen MECP2 (proteína de unión a metil-CpG 2).
Una mutación afecta a la producción de una proteína crítica para el desarrollo del cerebro
La copia mutada suele asociarse alALAmyloidosis alelo paterno, ya que se produce durante la espermatogénesis (solo las mujeres heredan el cromosoma X paterno, ya que el cromosoma Y paterno determina el sexo masculino del feto).
Las mujeres están afectadas casi exclusivamente.
Se cree que la mayoría de losLOSNeisseria varones con esta mutación mueren enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el útero o durante la primera infancia.
La incidencia es de 1 de cada 10 000 niñas a losLOSNeisseria 12 años enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum Estados Unidos.
Algunos niños pueden tener una mutación diferente, dando lugar a una forma menos destructiva del síndrome de Rett.
La esperanza de vida es mayor.
Menor riesgo de problemas intelectuales y del desarrollo
Presentación Clínica y Diagnóstico
Presentación clínica
LosLOSNeisseria individuos con síndrome de Rett suelen tener un desarrollo normal hasta losLOSNeisseria 7–24 meses de edad.
El estancamiento del desarrollo se produce entre losLOSNeisseria 6 y losLOSNeisseria 18 meses de edad.
Alrededor de losLOSNeisseria 1–4 años de edad, losLOSNeisseria individuos pierden las habilidades previamente adquiridas (regresión).
Signos y síntomas
Descripción
Retraso enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el crecimiento
El crecimiento del cerebro se enlentece después del nacimiento.
Microcefalia
Fracaso del crecimiento debido a las dificultades para masticar y tragar
Movimientos anormales
Reducción del control de la mano
Disminución de la capacidad de gatear o caminar
LosLOSNeisseria músculos pueden debilitarse (hipotonía, rigidez o espasticidad).
Movimientos repetitivos e inútiles de las manos (retorcerlas, apretarlas, aplaudirlas, golpearlas, frotarlas)
Movimientos oculares inusuales
Mirada intensa
Parpadeo excesivo
Estrabismo o cerrar un ojo a la vez
AtaxiaAtaxiaImpairment of the ability to perform smoothly coordinated voluntary movements. This condition may affect the limbs, trunk, eyes, pharynx, larynx, and other structures. Ataxia may result from impaired sensory or motor function. Sensory ataxia may result from posterior column injury or peripheral nerve diseases. Motor ataxia may be associated with cerebellar diseases; cerebral cortex diseases; thalamic diseases; basal ganglia diseases; injury to the red nucleus; and other conditions.Ataxia-telangiectasia de la marcha
Distonía (contracciones musculares sostenidas asociadas a movimientos y posturas incontroladas)
Exhalación forzada de aire o salivaSalivaThe clear, viscous fluid secreted by the salivary glands and mucous glands of the mouth. It contains mucins, water, organic salts, and ptyalin.Salivary Glands: Anatomy
Tragar aire (aerofagia)
Comportamientos anormales
LosLOSNeisseria lactantes son “plácidos”, exigen poca o ninguna atención de losLOSNeisseria padres
Expresiones faciales repentinas y extrañas
Largas carcajadas
Lamer la mano
Agarrar el pelo o la ropa
Cada vez más agitado, irritable e inquieto con la edad
El llanto inconsolable o losLOSNeisseria gritos son esporádicos.
Ataques de pánico
Discapacidades cognitivas
Pérdida de habilidades y funcionamiento intelectual
Pérdida de la capacidad de hablar
Algunos pueden experimentar una pérdida repentina del habla.
ApraxiaApraxiaA group of cognitive disorders characterized by the inability to perform previously learned skills that cannot be attributed to deficits of motor or sensory function. The two major subtypes of this condition are ideomotor and ideational apraxia, which refers to loss of the ability to mentally formulate the processes involved with performing an action. For example, dressing apraxia may result from an inability to mentally formulate the act of placing clothes on the body. Apraxias are generally associated with lesions of the dominant parietal lobe and supramarginal gyrus.Cranial Nerve Palsies (incapacidad de realizar movimientos aprendidos a la orden)
Otro
Huesos delgados y frágiles propensos a las fracturas
Latidos irregulares (pueden provocar la muerte súbita)
Escoliosis
Temblores y convulsiones (con trazo anormal del EEGEEGSeizures)
Diagnóstico
El diagnóstico se basa enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la presentación clínica, una toma de antecedentes clínicos detallada del paciente y una evaluación clínica exhaustiva.
Las pruebas genéticas para las mutaciones del gen MECP2pueden utilizarse para confirmar y/o descartar síndromes similares.
Imagen por Lecturio.
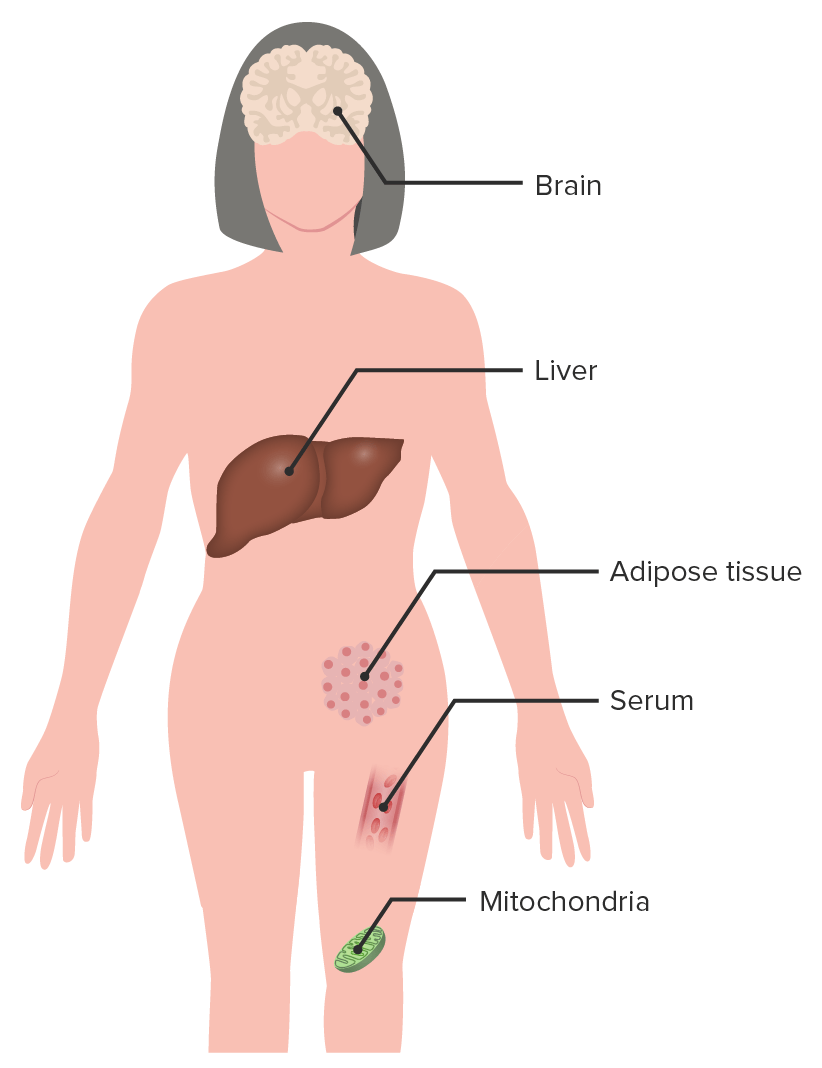
Clinical features/signs in RETT syndromeRett syndromeRett syndrome is a rare genetic neurological and developmental disorder that affects the development of the brain. There are various stages of Rett syndrome characterized by different symptoms and clinical signs, including increasing problems with the use of muscles that control movement, coordination, and communication. Rett Syndrome
BrainBrainThe part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem.Nervous System: Anatomy, Structure, and Classification:
Abnormalities in neurometabolites
Altered carbohydrate metabolism in CSF
Increased pyruvatePyruvateDerivatives of pyruvic acid, including its salts and esters.Glycolysis and lactate in CSF
Elevated lipidsLipidsLipids are a diverse group of hydrophobic organic molecules, which include fats, oils, sterols, and waxes.Fatty Acids and Lipids
Reduced choline phospholipid turnover
Oxidative changes
LiverLiverThe liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy:
InflammationInflammationInflammation is a complex set of responses to infection and injury involving leukocytes as the principal cellular mediators in the body’s defense against pathogenic organisms. Inflammation is also seen as a response to tissue injury in the process of wound healing. The 5 cardinal signs of inflammation are pain, heat, redness, swelling, and loss of function. Inflammation of the gallbladderGallbladderThe gallbladder is a pear-shaped sac, located directly beneath the liver, that sits on top of the superior part of the duodenum. The primary functions of the gallbladder include concentrating and storing up to 50 mL of bile. Gallbladder and Biliary Tract: Anatomy
Elevated triglyceridesTriglyceridesFatty Acids and Lipids (fatty liverLiverThe liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy)
Adipose tissueAdipose tissueAdipose tissue is a specialized type of connective tissue that has both structural and highly complex metabolic functions, including energy storage, glucose homeostasis, and a multitude of endocrine capabilities. There are three types of adipose tissue, white adipose tissue, brown adipose tissue, and beige or “brite” adipose tissue, which is a transitional form.Adipose Tissue: Histology:
Abnormal glucoseGlucoseA primary source of energy for living organisms. It is naturally occurring and is found in fruits and other parts of plants in its free state. It is used therapeutically in fluid and nutrient replacement.Lactose IntolerancetoleranceTolerancePharmacokinetics and Pharmacodynamics test
Insulin resistanceInsulin resistanceDiminished effectiveness of insulin in lowering blood sugar levels: requiring the use of 200 units or more of insulin per day to prevent hyperglycemia or ketosis.Diabetes Mellitus
Increased leptinLeptinA 16-kda peptide hormone secreted from white adipocytes. Leptin serves as a feedback signal from fat cells to the central nervous system in regulation of food intake, energy balance, and fat storage.Adipose Tissue: Histology, pyruvatePyruvateDerivatives of pyruvic acid, including its salts and esters.Glycolysis, and lactate
Decreased ghrelinGhrelinA 28-amino acid, Acylated, orexigenic peptide that is a ligand for growth hormone secretagogue receptors. Ghrelin is widely expressed but primarily in the stomach in the adults. Ghrelin acts centrally to stimulate growth hormone secretion and food intake, and peripherally to regulate energy homeostasis. Its large precursor protein, known as appetite-regulating hormone or motilin-related peptide, contains ghrelin and obestatin.Glucagonoma
Increase ammoniaAmmoniaA colorless alkaline gas. It is formed in the body during decomposition of organic materials during a large number of metabolically important reactions. Note that the aqueous form of ammonia is referred to as ammonium hydroxide.Acid-Base Balance
MitochondriaMitochondriaSemiautonomous, self-reproducing organelles that occur in the cytoplasm of all cells of most, but not all, eukaryotes. Each mitochondrion is surrounded by a double limiting membrane. The inner membrane is highly invaginated, and its projections are called cristae. Mitochondria are the sites of the reactions of oxidative phosphorylation, which result in the formation of ATP. They contain distinctive ribosomes, transfer RNAs; amino Acyl tRNA synthetases; and elongation and termination factors. Mitochondria depend upon genes within the nucleus of the cells in which they reside for many essential messenger RNAs. Mitochondria are believed to have arisen from aerobic bacteria that established a symbiotic relationship with primitive protoeukaryotes.The Cell: Organelles:
Abnormal mitochondrial structure
Altered function of the electron transport chainElectron transport chainThe electron transport chain (ETC) sends electrons through a series of proteins, which generate an electrochemical proton gradient that produces energy in the form of adenosine triphosphate (ATP).Electron Transport Chain (ETC)
Increase in oxidative stressOxidative stressA disturbance in the prooxidant-antioxidant balance in favor of the former, leading to potential damage. Indicators of oxidative stress include damaged DNA bases, protein oxidation products, and lipid peroxidation products.Cell Injury and Death
Estadios
Estadio I: Detención del desarrollo (6–18 meses de edad)
Puede presentarse con menos contacto visual y placidez
La consecución de losLOSNeisseria hitos del desarrollo se retrasa lentamente y cada vez más.
Estadio II: Deterioro rápido o regresión (1–4 años de edad)
LosLOSNeisseria niños pierden la capacidad de realizar habilidades previamente adquiridas.
La pérdida puede ser rápida o más gradual.
Reducción del crecimiento de la cabeza
Movimientos anormales de la mano (“retorcimiento”)
Hiperventilación
Gritar o llorar sin motivo aparente
Problemas de movimiento y coordinación
Pérdida de interacción social y comunicación
Estadio III: Meseta o pseudoestacionaria (2–10 años)
Se puede observar una regresión mínima de losLOSNeisseria síntomas con cierta mejora de las habilidades.
Las convulsiones pueden comenzar enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum esta etapa.
Estadio IV: Deterioro motorMotorNeurons which send impulses peripherally to activate muscles or secretory cells.Nervous System: Histology tardío (> 10 años)
Marcada por una movilidad reducida
Debilidad muscular
Contracturas articulares
Escoliosis
Complicaciones
LosLOSNeisseria problemas de sueño provocan una fatiga e inquietud importantes.
La dificultad para comer conduce a la desnutrición.
Problemas intestinales y de vejiga
Estreñimiento
Enfermedad por reflujo gastroesofágico (ERGE)
Incontinencia intestinal o urinaria
Enfermedad de la vesícula biliar
El dolorDolorInflammation puede acompañar a otros problemas, como problemas gastrointestinales o fracturas óseas.
Problemas musculares, óseos y articulares
Ansiedad y comportamiento problemático que puede dificultar el funcionamiento social
Necesidad de cuidados y asistencia de por vida para las actividades de la vida diaria
Duración de la vida acortada (la supervivencia media es de aproximadamente 45 años de edad)
Tratamiento
Trofinetida:
Primer tratamiento modificador de la enfermedad disponible
Mecanismo no completamente comprendido
Estudios demuestran mejoría a corto plazo; aún no se haHAHemolytic anemia (HA) is the term given to a large group of anemias that are caused by the premature destruction/hemolysis of circulating red blood cells (RBCs). Hemolysis can occur within (intravascular hemolysis) or outside the blood vessels (extravascular hemolysis).Hemolytic Anemia establecido la mejoría a largo plazo.
Control estricto de:
Talla
Peso
Control y optimización nutricional
Detección y tratamiento de complicaciones (e.g., ERGE, densidad ósea, convulsiones).
Terapias:
Físicas
Ocupacional
Comunicación
Diagnóstico Diferencial
Las siguientes afecciones son diagnósticos diferenciales para el síndrome de Rett:
Síndrome de Angelman: una enfermedad autosómica del neurodesarrollo resultante de la impronta genómica epigenética específica del sexo y la disomía uniparental del cromosoma 15 paterno con una pérdida funcional simultánea de la parte materna. Se caracteriza por una risa inapropiada, un comportamiento aparentemente alegre, poca capacidad de atención, falta de habla adecuada, uso de habilidades de comunicación no verbal, ataxiaAtaxiaImpairment of the ability to perform smoothly coordinated voluntary movements. This condition may affect the limbs, trunk, eyes, pharynx, larynx, and other structures. Ataxia may result from impaired sensory or motor function. Sensory ataxia may result from posterior column injury or peripheral nerve diseases. Motor ataxia may be associated with cerebellar diseases; cerebral cortex diseases; thalamic diseases; basal ganglia diseases; injury to the red nucleus; and other conditions.Ataxia-telangiectasia, temblores finos, movimientos espasmódicos y un grave retraso enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el desarrollo.
Síndrome de Prader-Willi: una rara enfermedad del neurodesarrollo autosómica resultante de la impronta genómica y la disomía uniparental del cromosoma 15 materno con una pérdida funcional simultánea de la parte parental. Se caracteriza por hipotonía, letargo, dificultades de alimentación, dificultades respiratorias e hipogonadismo alALAmyloidosis nacer; con escoliosis, estrabismo, retraso del habla, retraso intelectual y problemas de comportamiento durante la infancia.
Síndrome de Landau-Kleffner: un raro trastorno del neurodesarrollo caracterizado por la pérdida de comprensión del lenguaje, afasia y convulsiones, con hallazgos de electroencefalograma (EEGEEGSeizures) gravemente anormales durante el sueño y convulsiones.
Trastorno del espectro autista: trastorno del neurodesarrollo de aparición temprana enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la infancia, caracterizado por escasas habilidades sociales, restricción de la interacción social y la comunicación, y comportamientos repetitivos y estereotipados. Dado que se trata de un espectro, algunos pacientes presentan un deterioro del lenguaje y de losLOSNeisseria niveles intelectuales, mientras que otros pueden tener niveles normales o incluso avanzados.
Parálisis cerebral: un síndrome de discapacidad motora causado por una lesión no progresiva del sistema nervioso central. Clasificados según el tono muscular, la distribución y el momento presumible de la lesión. También puede presentar alteraciones sensoriales, cognitivas, de comunicación y de comportamiento; epilepsia y alteraciones musculoesqueléticas.
Trastorno por deficiencia de CDKL5 (quinasa dependiente de ciclina 5): trastorno genético poco frecuente que se hereda con un patrón ligado alALAmyloidosis cromosoma X y que se caracteriza por convulsiones de aparición temprana y graves alteraciones del neurodesarrollo. También puede presentar escoliosis, problemas visuales, problemas sensoriales y diversas dificultades gastrointestinales.
Trastornos relacionados con FOXG1 o síndrome de FOXG1: grupo de trastornos causados por una mutación del gen FOXG1. LosLOSNeisseria individuos afectados se desarrollan normalmente enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el periodo perinatal. A continuación, se produce una microcefalia progresiva, convulsiones, retrasos enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el desarrollo y discapacidad intelectual grave. También puede presentar estreñimiento, reflujo gastroesofágico, escoliosis, deformidades de losLOSNeisseria pies y movimientos estereotipados de las manos.
Referencias
Porter, R. S., Kaplan, J. L., Lynn, R. B., & Reddy, M. T. (Eds.). (2018). The Merck manual of diagnosis and therapy (20th ed.). Merck Sharp & Dohme Corp.
Neul, J. L., & Kaufmann, W. E. (2023). Rett syndrome: Clinical diagnosis and management. Nature Medicine, 29(5), 953-960. https://doi.org/10.1038/s41591-023-02398-1
Pini, G., & Giannini, C. (2022). Genetics of Rett Syndrome: Mechanisms and Implications (2nd ed.). Springer.
Sellars, E. A., & Hopkin, R. J. (2021). Pediatric Genetics (3rd ed.). Springer.
Zeldin, R. K., & Schwartz, M. (2022). Immunology Made Ridiculously Simple. MedMaster Inc.
Halbach, N. S. J., Smeets, E. E. J., van den Braak, N., et al. (2012). Genotype-phenotype relationships as prognosticators in Rett syndrome should be handled with care in clinical practice. American Journal of Medical Genetics – Part A, 158(2), 340–350.https://doi.org/10.1002/ajmg.a.34418
Obtenga Medical Premium para poner a prueba sus conocimientos
Lecturio Medical Premium le brinda acceso completo a todo el contenido y las funciones
Obtenga Premium para ver todos los vídeos
Verifica tu correo electrónico para obtener una prueba gratuita.
Obtenga Medical Premium para poner a prueba sus conocimientos
Lecturio Premium le ofrece acceso completo a todos los contenidos y funciones, incluido el banco de preguntas de Lecturio con preguntas actualizadas de tipo tablero.