Cri du chat is the French term for "cat-cry" or "call of the cat." The term refers to the cat-like cry of a pediatric patient with cri-du-chat syndrome. The condition is a rare genetic disorder caused by deletion mutations on chromosome Chromosome In a prokaryotic cell or in the nucleus of a eukaryotic cell, a structure consisting of or containing DNA which carries the genetic information essential to the cell. Basic Terms of Genetics 5. Cri-du-chat syndrome is more common in females than in males. Aside from the characteristic cry, the condition also presents with dysphagia Dysphagia Dysphagia is the subjective sensation of difficulty swallowing. Symptoms can range from a complete inability to swallow, to the sensation of solids or liquids becoming "stuck." Dysphagia is classified as either oropharyngeal or esophageal, with esophageal dysphagia having 2 sub-types: functional and mechanical. Dysphagia, low birth weight, poor growth, and severe cognitive, speech, and motor Motor Neurons which send impulses peripherally to activate muscles or secretory cells. Nervous System: Histology disabilities.
Last updated: Dec 15, 2025
Symptoms vary and depend on the amount of deleted genetic material.
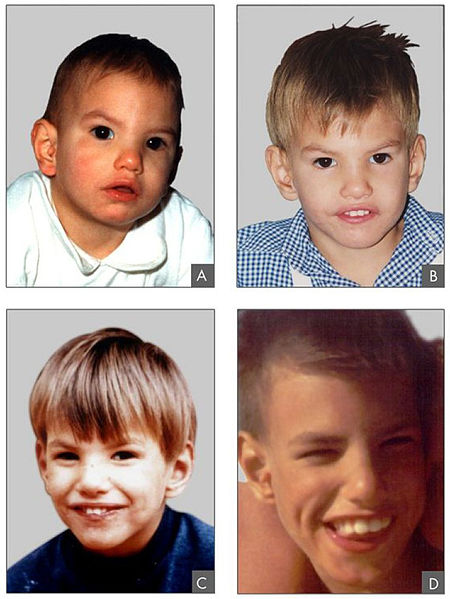
Phenotypic features associated with cri-du-chat syndrome
Image: “Criduchat” by Paola Cerruti Mainardi. License: CC BY 2.0The following conditions are differential diagnoses of cri-du-chat syndrome: