Congenital renal abnormalities arise from embryologic/genetic defects and cause a variety of isolated or syndromic renal disorders, including renal agenesis Agenesis Teratogenic Birth Defects, dysgenesis, and ectopia. Congenital renal abnormalities are generally identified prenatally and represent approximately ⅓ of all prenatal anomalies. Because of the fetal kidney’s role in the production of amniotic fluid Amniotic fluid A clear, yellowish liquid that envelopes the fetus inside the sac of amnion. In the first trimester, it is likely a transudate of maternal or fetal plasma. In the second trimester, amniotic fluid derives primarily from fetal lung and kidney. Cells or substances in this fluid can be removed for prenatal diagnostic tests (amniocentesis). Placenta, Umbilical Cord, and Amniotic Cavity, oligohydramnios Oligohydramnios Oligohydramnios refers to amniotic fluid volume less than expected for the current gestational age. Oligohydramnios is diagnosed by ultrasound and defined as an amniotic fluid index (AFI) of ‰¤ 5 cm or a single deep pocket (SDP) of < 2 cm in the 2nd or 3rd trimester. Oligohydramnios detected on prenatal ultrasounds often prompts the workup that identifies congenital renal anomalies. Unilateral renal involvement in the presence of a functioning contralateral kidney may only be an incidental finding later in life. In many cases, treatment is supportive.
Last updated: Dec 15, 2025
The kidney develops in the pelvis Pelvis The pelvis consists of the bony pelvic girdle, the muscular and ligamentous pelvic floor, and the pelvic cavity, which contains viscera, vessels, and multiple nerves and muscles. The pelvic girdle, composed of 2 “hip” bones and the sacrum, is a ring-like bony structure of the axial skeleton that links the vertebral column with the lower extremities. Pelvis: Anatomy and migrates cranially. Three separate renal systems form in sequence, giving rise to the kidney, in association with the urinary tract Urinary tract The urinary tract is located in the abdomen and pelvis and consists of the kidneys, ureters, urinary bladder, and urethra. The structures permit the excretion of urine from the body. Urine flows from the kidneys through the ureters to the urinary bladder and out through the urethra. Urinary Tract: Anatomy and urogenital system:
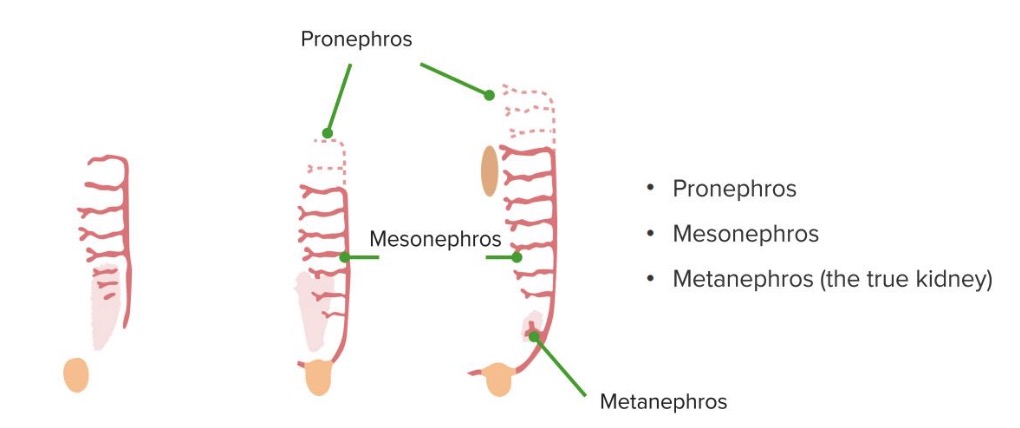
The 3 phases of kidney development
Image by Lecturio.
Metanephros
Image by Lecturio.Renal embryologic disorders affecting the size, the shape, or the structure of kidney parenchyma (renal dysgenesis):
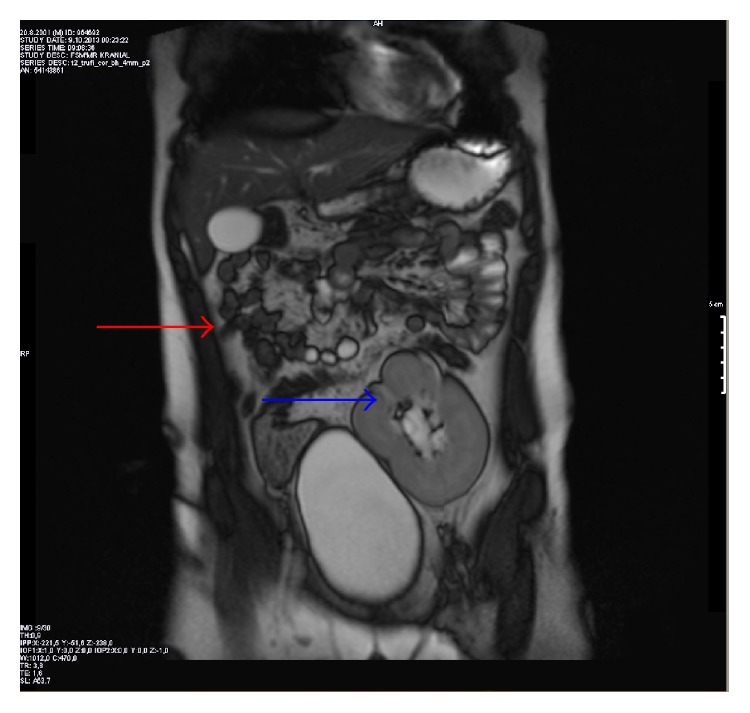
Incidental discovery of renal agenesis in a 12-year-old boy: Magnetic resonance imaging (MRI) of the abdomen on the coronal plane shows right renal agenesis (red arrow) and left ectopic pelvic kidney over the urinary bladder (blue arrow).
Image: “Figure 2” by Altun et al. License: CC BY 2.0.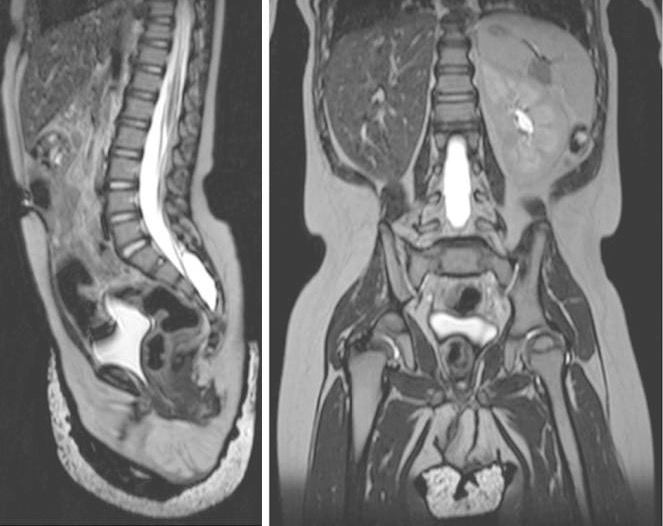
Sagittal (left) and coronal (right) T2-weighted MRI views of the abdomen and pelvis, showing right renal agenesis and complete absence of Müllerian/paramesonephric duct structures in an 18-month-old infant
Image: “Figure 2” by William Mifsud et al. License: CC BY 3.0.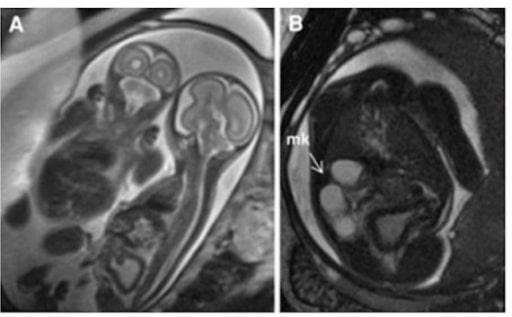
Multicystic kidney: T2-weighted HASTE imaging of monochorionic, monoamniotic twins. Panel A demonstrates that the affected twin is on the right with a thickened and distended bladder.
Panel B is an axial slice demonstrating a multicystic kidney (arrow).
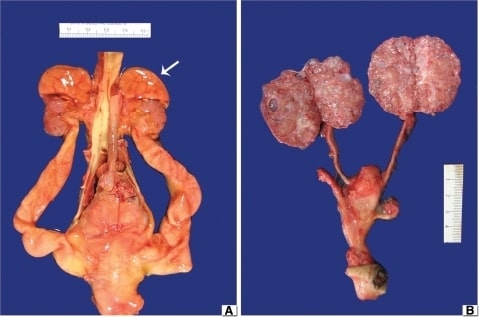
(A) Obstructive MCDK: Notice the hypoplastic kidneys with the adrenal glands still at the upper poles (arrow). The aorta is centrally located and there are bilateral hydroureters.
(B) Nonobstructive MCDK: The ureters are unremarkable and portions of the two umbilical arteries run parallel to the urinary bladder. The kidneys are bi-valved to demonstrate numerous small cysts scattered through the cortex and medulla.
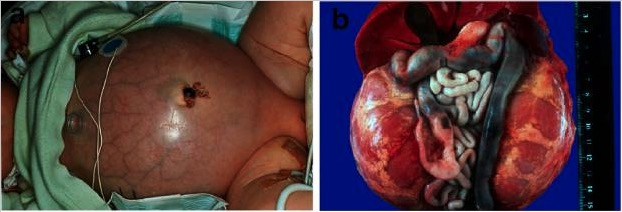
Autosomal recessive polycystic kidney disease.
A: Baby with a distended abdomen due to voluminous kidneys that led to respiratory problems and early demise.
B: Abdominal situs of a perinatally demised ARPKD patient with symmetrically enlarged kidneys that maintained their reniform configuration.
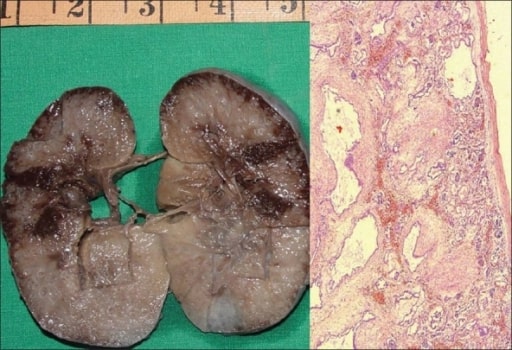
Autosomal recessive polycystic kidney disease in a fetus.
A: Gross specimen of the right kidney measuring 10 × 7 × 4 cm. The cut surface is spongy with poor corticomedullary differentiation. There are multiple, tiny cysts with some at right angles to the cortical surface.
B: Photomicrograph showing numerous cysts lined by a single layer of low cuboidal epithelial cells with thick peritubular mesenchyme. The glomeruli are normal (H and E, ×40).
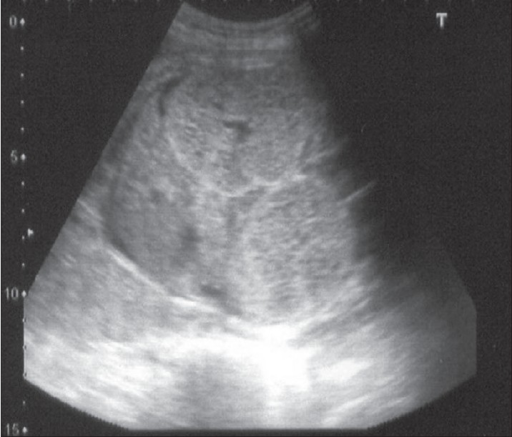
Antenatal ultrasound at 24 weeks showing bilateral, symmetrically enlarged, echogenic kidneys filling the fetal abdomen. The liver is normal.
Image: “F0001” by the Department of Urology, Kasturba Medical College, Manipal, India. License: CC BY 2.0.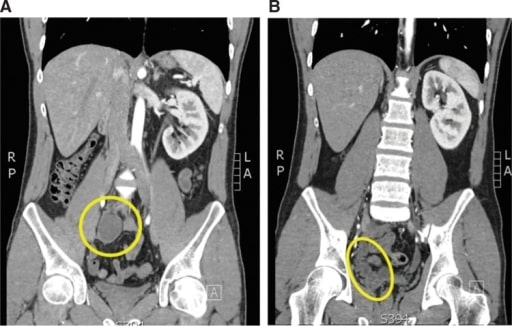
A 24-year-old man with recurrent epididymitis and a ureteral insertion into the seminal vesicle. A: A coronal cut of the computed tomography (CT) scan showing an atrophic right pelvic kidney (yellow circle). B: On this coronal cut of the CT scan, the ureter and seminal vesicle complex can be seen (yellow ellipse).
Image: “Atrophic and ectopic right kidney” by the U.S. National Library of Medicine. License: CC BY 4.0.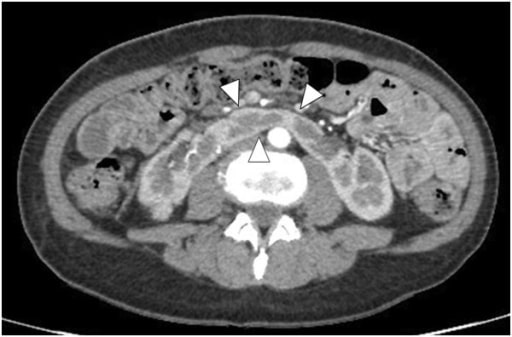
Horseshoe kidney on contrast-enhanced CT: Arrowheads show the isthmus.
Image: “Enhanced abdominal computed tomography” by the Department of Urology, Hirosaki University Graduate School of Medicine, 5 Zaifucho, Hirosaki 036-8562, Japan. License: CC BY 2.0.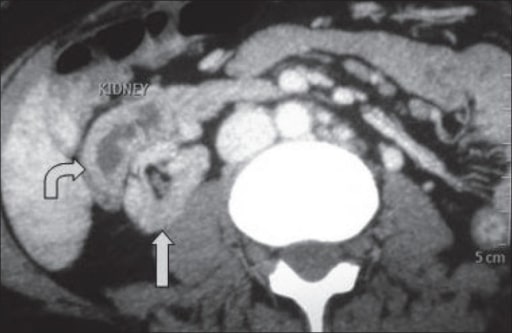
A CT showing crossed fused ectopia: The ectopic kidney is situated anterolateral to the orthotopic kidney.
Image: “A computed tomography showing crossed fused ectopia” by the Department of Urology and Transplantation Surgery, Institute of Kidney Diseases and Research Centre, Institute of Transplantation Sciences, Civil Hospital Campus, Asarwa, Ahmedabad, Gujarat, India. License: CC BY 2.0.