


As anomalias renais congénitas surgem de defeitos embriológicos/genéticos e causam uma variedade de distúrbios renais isolados ou sindrómicos, incluindo agenesia renal, disgenesia e ectopia. As anomalias renais congénitas são geralmente identificadas no período pré-natal e representam, aproximadamente, ⅓ de todas as anomalias pré-natais. Devido ao importante papel do rim fetal na produção de líquido amniótico, se detetado oligoâmnios em ultrassonografias pré-natais, geralmente, despoleta-se investigação adicional que identifica anomalias renais congénitas. O envolvimento renal unilateral, na presença de um rim contralateral funcionante, pode ser apenas um achado incidental mais MAIS Androgen Insensitivity Syndrome tarde na vida. Em muitos casos, o tratamento é de suporte.
Last updated: Dec 15, 2025
O rim desenvolve-se na pelve e migra cranialmente. Formam-se três sistemas renais separados, em sequência, dando origem ao rim, em associação com o trato urinário e o sistema urogenital:
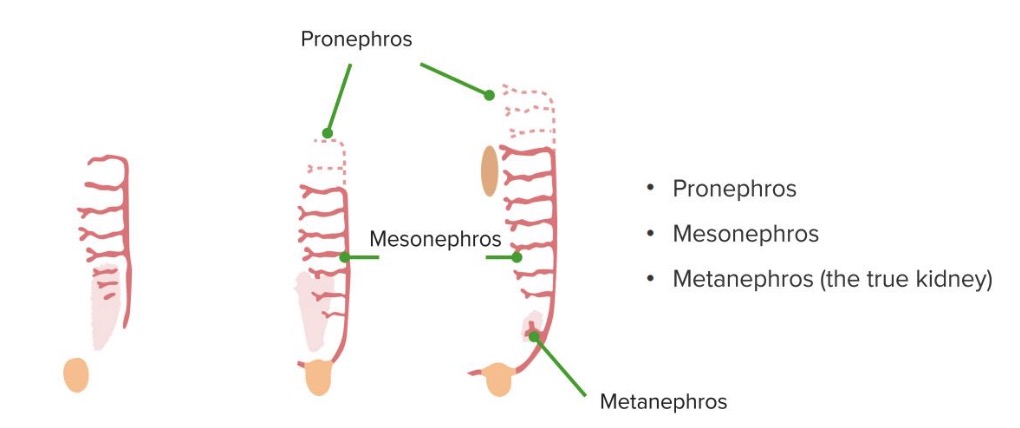
As 3 fases do desenvolvimento renal
Imagem por Lecturio.
Metanefros
Imagem por Lecturio.Distúrbios embriológicos renais que afetam o tamanho, a forma ou a estrutura do parênquima renal (disgenesia renal):
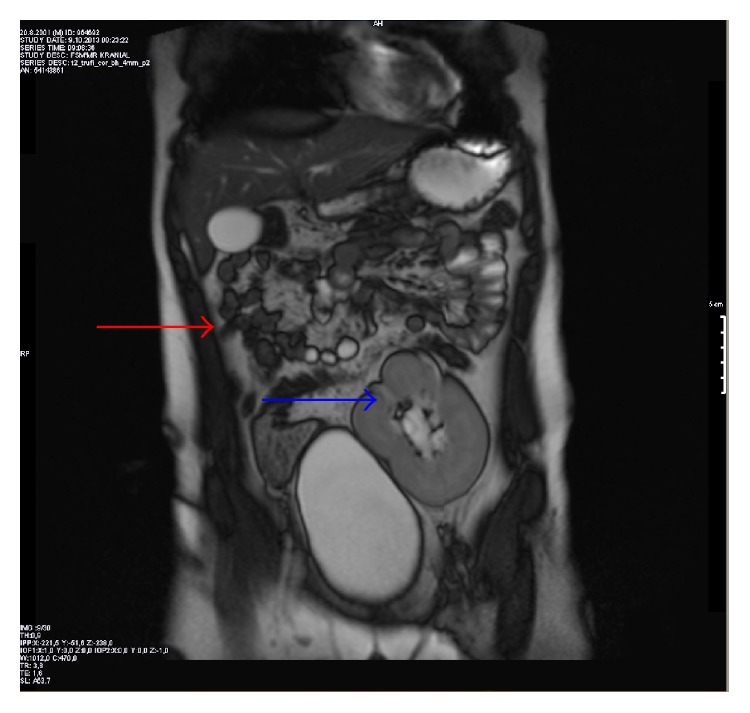
Descoberta incidental de agenesia renal num menino de 12 anos: A ressonância magnética (RMN) do abdómen, no plano coronal, mostra agenesia renal direita (seta vermelha) e um rim pélvico ectópico esquerdo sobre a bexiga urinária (seta azul).
Imagem: “Figure 2” por Altun et al. Licença: CC BY 2.0.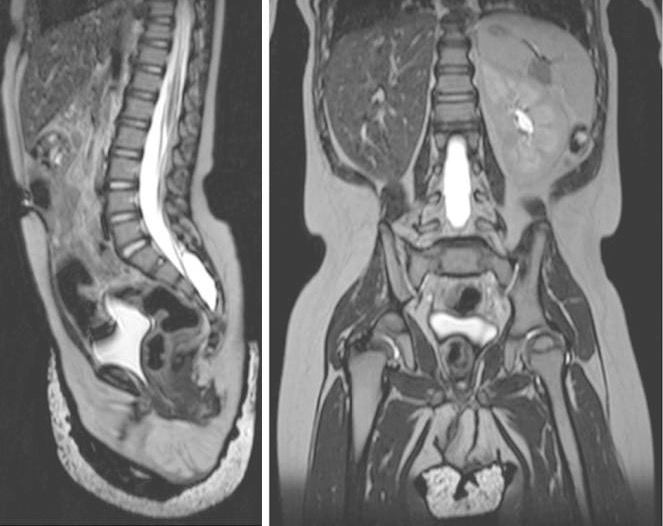
Imagens de RMN ponderada em T2, em corte sagital (esquerda) e coronal (direita), do abdómen e da pelve, a demonstrar agenesia renal direita e ausência completa de estruturas dos ductos müllerianos/paramesonéfricos num lactente de 18 meses
Imagem: “Figure 2” por William Mifsud et al. Licença: CC BY 3.0.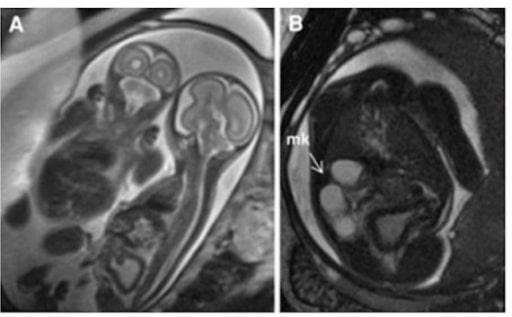
Rim multiquístico: imagem HASTE ponderada em T2 de gémeos monocoriónicos e monoamnióticos. O painel A demonstra que o gémeo afetado está à direita com uma bexiga espessada e distendida.
O painel B é um corte axial a demonstrar um rim multiquístico (seta).
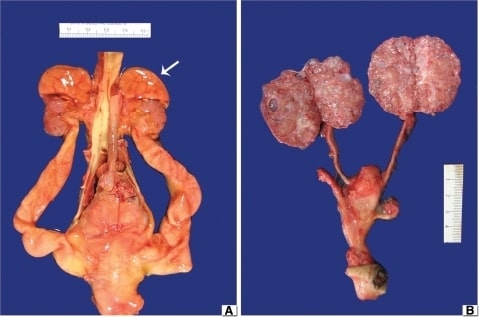
(A) MCDK obstrutivo: Observar os rins hipoplásicos com as glândulas suprarrenais ainda nos polos superiores (seta). A aorta está localizada centralmente e visualizam-se hidroureteres bilaterais.
(B) MCDK não obstrutivo: Os ureteres são normais e porções das duas artérias umbilicais correm paralelamente à bexiga urinária. Os rins estão divididos para demonstrar numerosos pequenos quistos espalhados pelo córtex e pela medula.
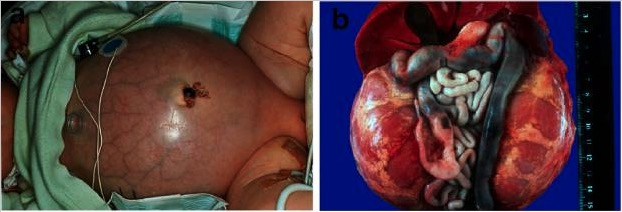
Doença renal poliquística autossómica recessiva.
R: Bebé com abdómen distendido pela presença de rins volumosos, que levaram a problemas respiratórios e morte precoce.
B: Abdómen de um doente que sofreu morte perinatal por ARPKD, com rins simetricamente aumentados que mantiveram a sua configuração reniforme.
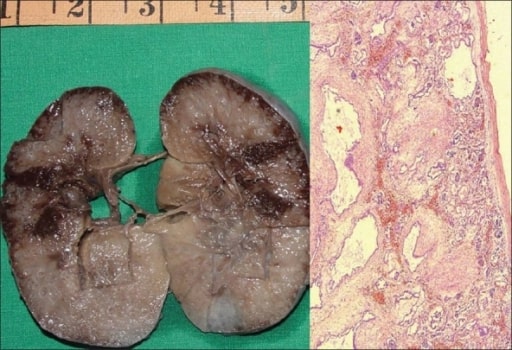
Doença renal poliquística autossómica recessiva num feto.
A: Amostra macroscópica do rim direito a medir 10 × 7 × 4 cm. A superfície de corte é esponjosa com baixa diferenciação corticomedular. Existem vários quistos minúsculos, alguns em ângulo reto com a superfície cortical.
B: Fotomicrografia a demonstrar quistos numerosos revestidos por uma única camada de células epiteliais cuboidais baixas, com mesênquima peritubular espesso. Os glomérulos são normais (H e E, ×40).
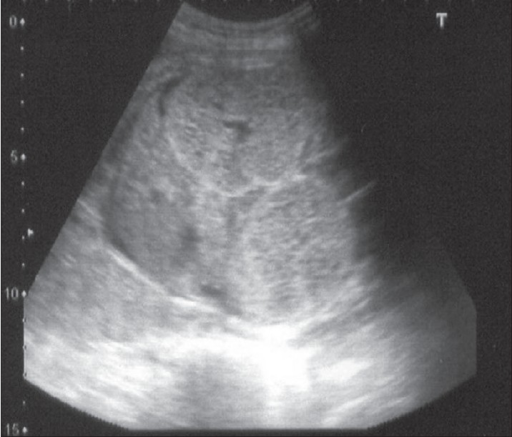
Ecografia pré-natal, às 24 semanas, a demonstrar rins simetrica e bilateralmente aumentados e ecogénicos a preencher o abdómen fetal. O fígado está normal.
Imagem : “F0001” por the Department of Urology, Kasturba Medical College, Manipal, India. Licença: CC BY 2.0.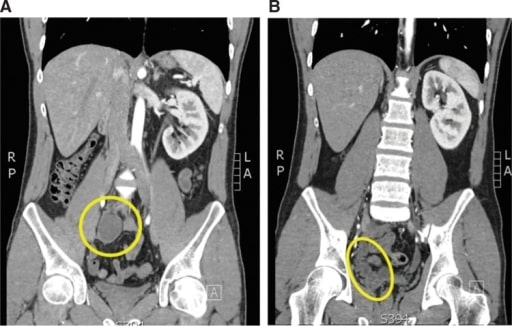
Homem de 24 anos com epididimite recorrente e inserção ureteral na vesícula seminal. A: Corte coronal de tomografia computadorizada (TC) a demonstrar um rim pélvico direito atrófico (círculo amarelo). B: Neste corte coronal da TC, observa-se o complexo ureteral e da vesícula seminal (elipse amarela).
Imagem: “Atrophic and ectopic right kidney” por the U.S. National Library of Medicine. Licença: CC BY 4.0.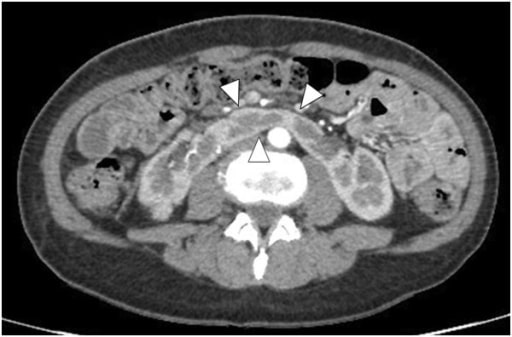
Rim em ferradura na TC com contraste: As pontas de seta mostram o istmo.
Imagem: “Enhanced abdominal computed tomography” por the Department of Urology, Hirosaki University Graduate School of Medicine, 5 Zaifucho, Hirosaki 036-8562, Japan. Licença: CC BY 2.0.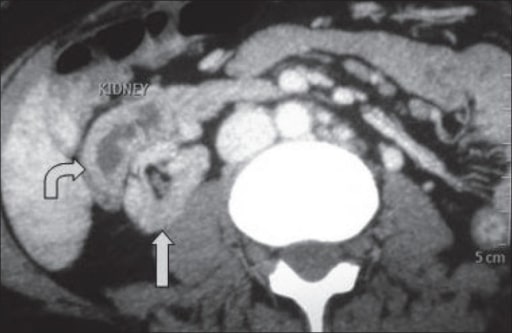
Uma TC a demonstrar ectopia fundida cruzada: O rim ectópico está situado anterolateralmente ao rim ortotópico.
Imagem: “A computed tomography showing crossed fused ectopia” por the Department of Urology and Transplantation Surgery, Institute of Kidney Diseases and Research Centre, Institute of Transplantation Sciences, Civil Hospital Campus, Asarwa, Ahmedabad, Gujarat, India. Licença: CC BY 2.0.