


Cri du chat é um termo francês para "choro de gato" ou "chamamento do gato". O termo refere-se ao choro semelhante ao de um gato de um doente pediátrico com síndrome de cri-du-chat. A condição é um distúrbio genético raro causado por mutações de deleção no cromossoma 5. A síndrome de Cri-du-chat é mais MAIS Androgen Insensitivity Syndrome comum em mulheres do que em homens. Além do choro característico, a condição também se apresenta com disfagia, baixo peso ao nascimento, défice de crescimento e défices cognitivos graves da fala e motores.
Last updated: Dec 15, 2025
Os sintomas variam e dependem da quantidade de material genético deletado.
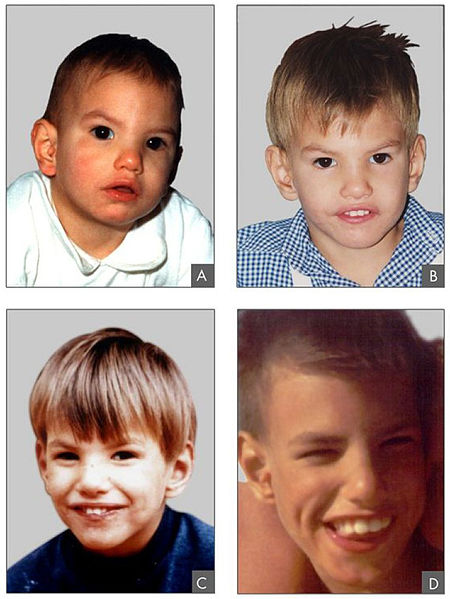
Características fenotípicas associadas à síndrome de cri-du-chat
Imagem: “Criduchat” por Paola Cerruti Mainardi. Licença: CC BY 2.0As seguintes condições são diagnósticos diferenciais da síndrome cri-du-chat:
