Cholestasis in neonates and young infants is conjugated hyperbilirubinemia in the 1st 3 months of life due to impaired bile excretion. Biliary tract malformations involving the gallbladder and bile duct are grouped into cystic and noncystic obliterative cholangiopathies, the most common of which is biliary atresia. Less common causes include the genetic Alagille syndrome, infectious causes, and metabolic disorders. Clinical presentation is with obstructive jaundice. Ultrasonography and MRCP are useful diagnostic tools, and a prenatal diagnosis is sometimes made with ultrasonography. Cystic causes frequently require surgery to correct the defect and allow for normal child growth. Liver transplantation may be needed in cases of biliary atresia with portal hypertension.
Last updated: Feb 16, 2026
Cholestasis in neonates and young infants is conjugated hyperbilirubinemia Conjugated hyperbilirubinemia Hyperbilirubinemia of the Newborn in the 1st 3 months of life due to impaired bile Bile An emulsifying agent produced in the liver and secreted into the duodenum. Its composition includes bile acids and salts; cholesterol; and electrolytes. It aids digestion of fats in the duodenum. Gallbladder and Biliary Tract: Anatomy excretion.
Biliary tract Biliary tract Bile is secreted by hepatocytes into thin channels called canaliculi. These canaliculi lead into slightly larger interlobular bile ductules, which are part of the portal triads at the “corners” of hepatic lobules. The bile leaves the liver via the right and left hepatic ducts, which join together to form the common hepatic duct. Gallbladder and Biliary Tract: Anatomy malformations involving the gallbladder Gallbladder The gallbladder is a pear-shaped sac, located directly beneath the liver, that sits on top of the superior part of the duodenum. The primary functions of the gallbladder include concentrating and storing up to 50 mL of bile. Gallbladder and Biliary Tract: Anatomy and bile Bile An emulsifying agent produced in the liver and secreted into the duodenum. Its composition includes bile acids and salts; cholesterol; and electrolytes. It aids digestion of fats in the duodenum. Gallbladder and Biliary Tract: Anatomy duct are grouped into cystic Cystic Fibrocystic Change and noncystic obliterative cholangiopathies, the most common of which is biliary atresia Atresia Hypoplastic Left Heart Syndrome (HLHS). Less common causes include the genetic Alagille syndrome Alagille Syndrome Hyperbilirubinemia of the Newborn, infectious causes, and metabolic disorders.
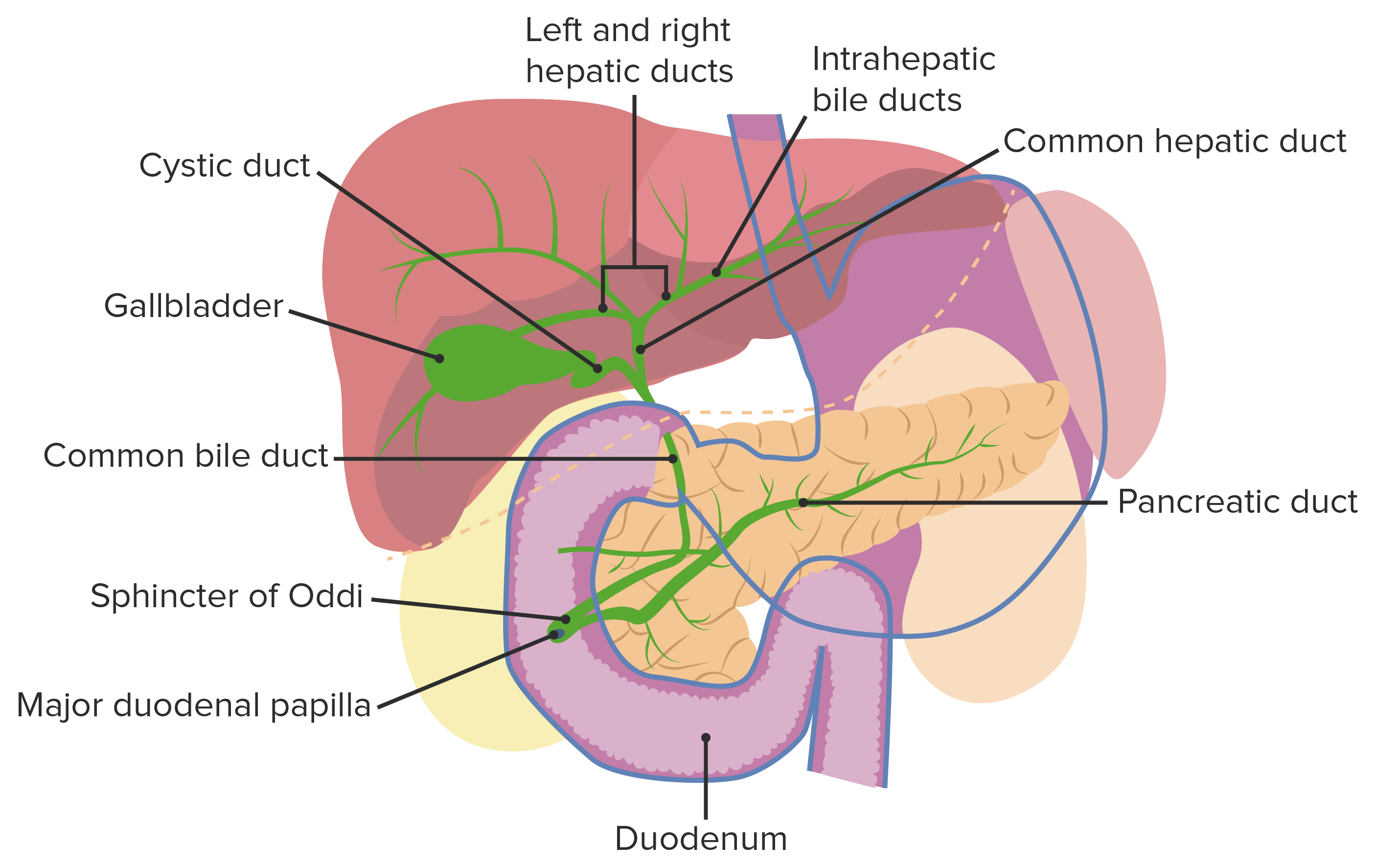
Normal biliary tract anatomy
Image by Lecturio.Biliary atresia Atresia Hypoplastic Left Heart Syndrome (HLHS) is an idiopathic Idiopathic Dermatomyositis, progressive, fibrotic obliteration of the extrahepatic biliary tree Biliary tree The bile ducts and the gallbladder. Gallbladder and Biliary Tract: Anatomy that presents with biliary obstruction within the 1st 3 months of life.
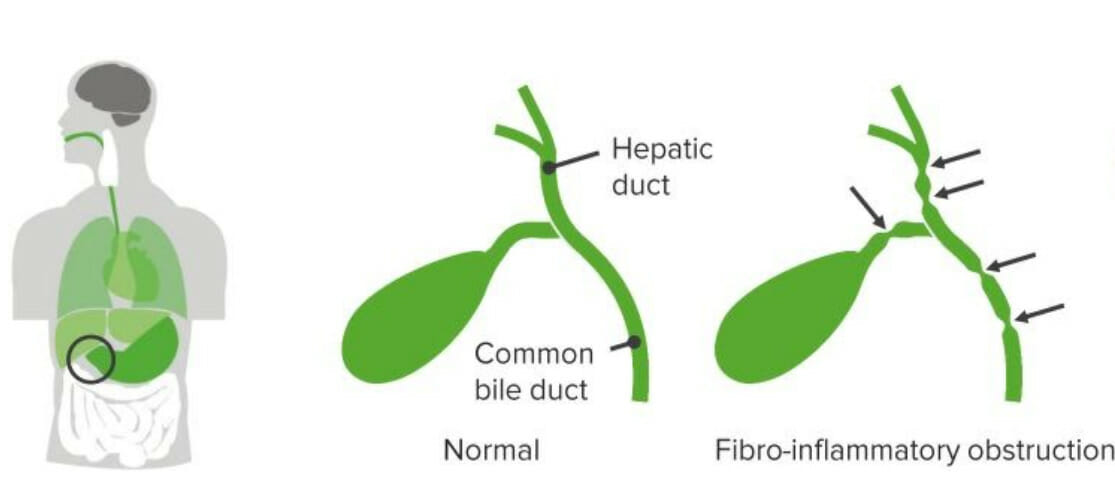
Biliary atresia
Image by Lecturio.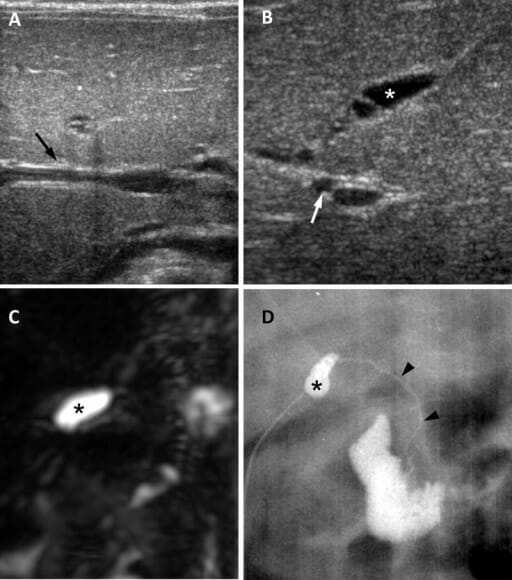
Biliary atresia in a 65-day-old girl:
A: Ultrasound in the transverse plane shows a negative triangular cord sign (arrow).B: Ultrasound image in oblique subcostal plane shows atretic gallbladder measuring 0.8 cm (asterisk) and enlarged hepatic artery measuring 1.5 mm (arrow).
C: 3-dimensional MRCP image shows no visible extrahepatic biliary tree and a small gallbladder (asterisk).
D: Surgical cholangiography shows small gallbladder (asterisk) and a patent but extremely hypoplastic common bile duct (arrowheads).
Image: “Biliary atresia in a 65-day-old girl” by Department of Radiology and Center for Imaging Science, Samsung Medical Center, Sungkyunkwan University School of Medicine. License: CC BY 4.0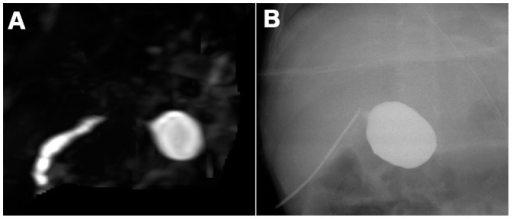
A: Biliary atresia in a 90-day-old female infant. 3-dimensional-MRCP does not display the extrahepatic biliary ducts except for the cystic common bile duct.
B: Intraoperative cholangiography in the same infant.
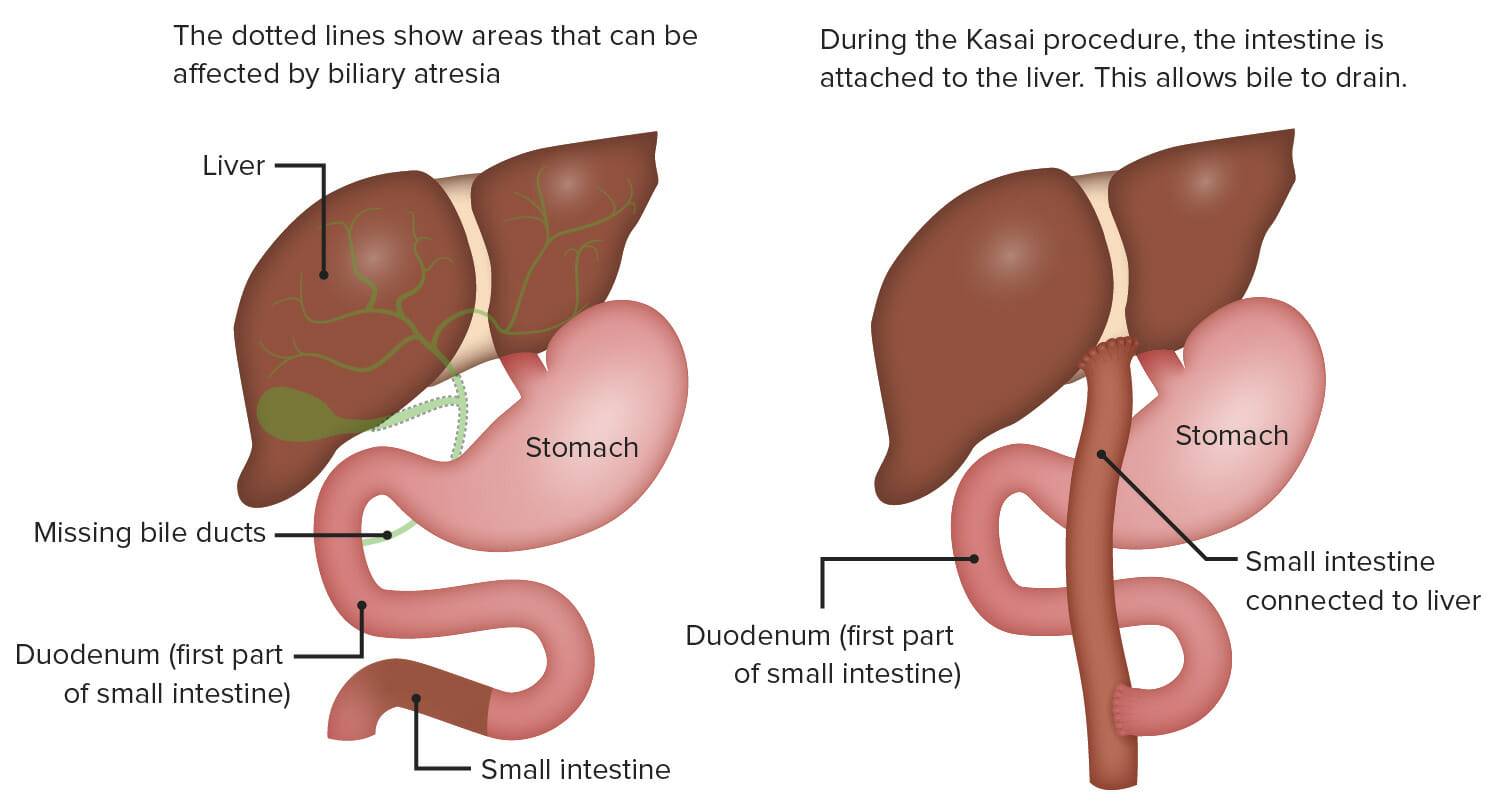
Portoenterostomy (Kasai procedure):
This surgery involves exposing the porta hepatis by radical excision of all bile duct tissue up to the liver capsule and attaching a Roux-en-Y loop of jejunum to the exposed liver capsule above the bifurcation of the portal vein.
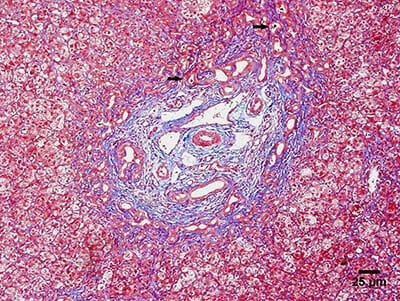
Photomicrograph showing the main histologic findings of biliary atresia:
Liver biopsy showing ductular proliferation and biliary plugs (arrows).
Alagille syndrome Alagille Syndrome Hyperbilirubinemia of the Newborn is a genetic disorder characterized by a paucity of bile Bile An emulsifying agent produced in the liver and secreted into the duodenum. Its composition includes bile acids and salts; cholesterol; and electrolytes. It aids digestion of fats in the duodenum. Gallbladder and Biliary Tract: Anatomy ducts in the liver Liver The liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy, leading to cholestasis. This syndrome also affects other organs.
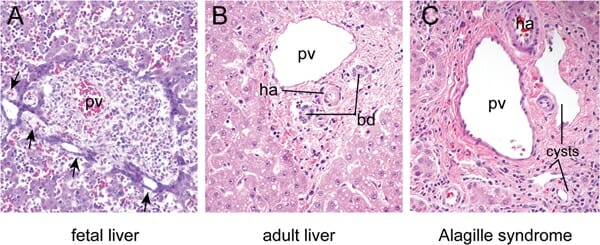
A: Fetal liver at 16 weeks of development showing focal dilations (arrows) in the biliary epithelial cell precursors surrounding the portal vein (PV).
B: Mature liver showing bile ducts (BD) and hepatic artery (HA) embedded in the periportal mesenchyme in a characteristic arrangement known as the “portal triad.” Hepatocytes arranged in chords surround the portal triad.
C: The portal triad region from an individual with Alagille syndrome shows ductal cysts rather than normal bile ducts due to defects in ductal plate remodeling.
Choledochal cysts Cysts Any fluid-filled closed cavity or sac that is lined by an epithelium. Cysts can be of normal, abnormal, non-neoplastic, or neoplastic tissues. Fibrocystic Change are congenital bile Bile An emulsifying agent produced in the liver and secreted into the duodenum. Its composition includes bile acids and salts; cholesterol; and electrolytes. It aids digestion of fats in the duodenum. Gallbladder and Biliary Tract: Anatomy duct anomalies that cause abnormal enlargement and obstruction of the intrahepatic and/or extrahepatic bile Bile An emulsifying agent produced in the liver and secreted into the duodenum. Its composition includes bile acids and salts; cholesterol; and electrolytes. It aids digestion of fats in the duodenum. Gallbladder and Biliary Tract: Anatomy ducts.
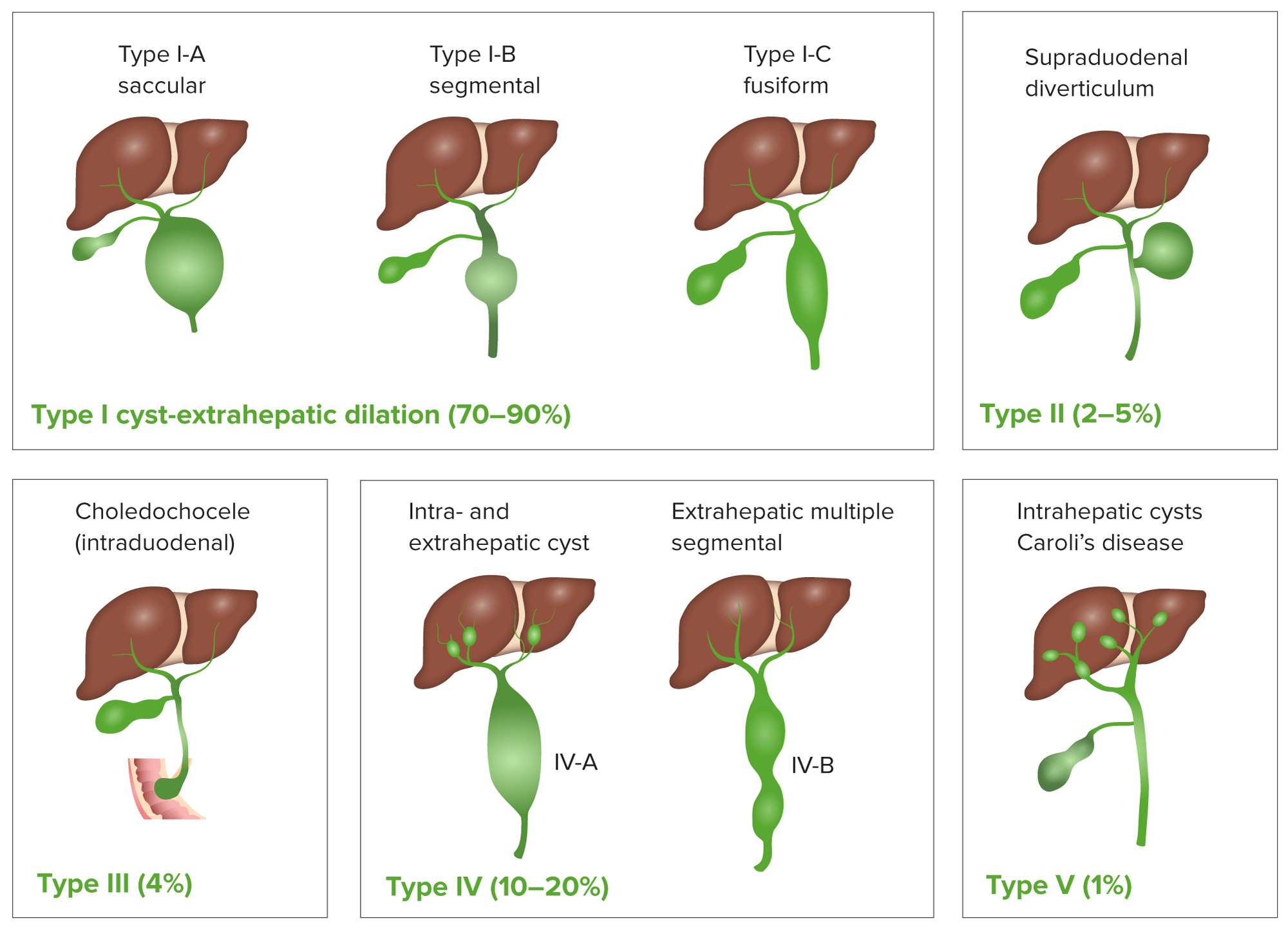
Classification of choledochal cyst
Image by Lecturio.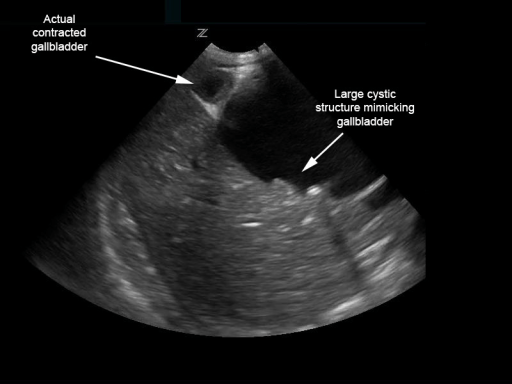
Biliary choledochal cyst mimicking distended gallbladder with multiple stones:
However, the contracted gallbladder is adjacent.
Image: “Biliary choledochal cyst” by University of Massachusetts, Department of Emergency Medicine, Boston. License: CC BY 4.0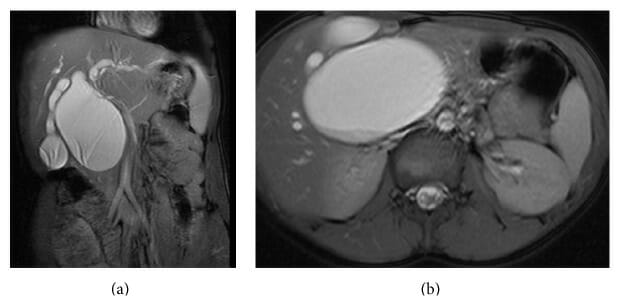
(a): MRCP showing type I-A saccular choledochal cyst.
(b): MRCP (cross section) showing large type I-A saccular choledochal cyst.
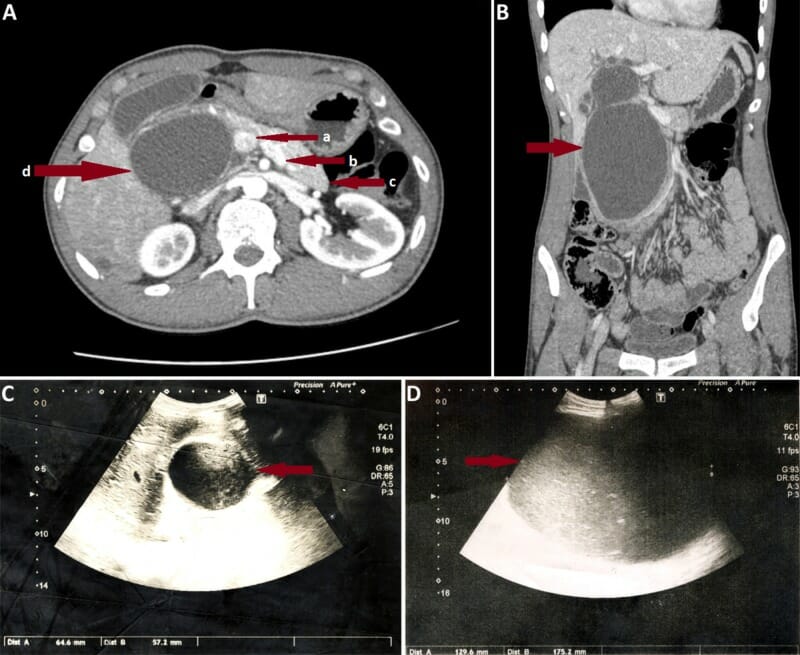
A: Coronal section of abdominal CT scan in an individual with a large type I choledochal cyst. Red arrows show portal vein (a), splenic vein (b), pancreatic body and tail (c), and giant choledochal cyst (d), which obliterates the pancreatic head.
B: Axial CT scan of the same individual.
C: Ultrasound image in the same individual on the day of admission. Note the rapid increase in cyst size observed on the 7th day of admission (D).
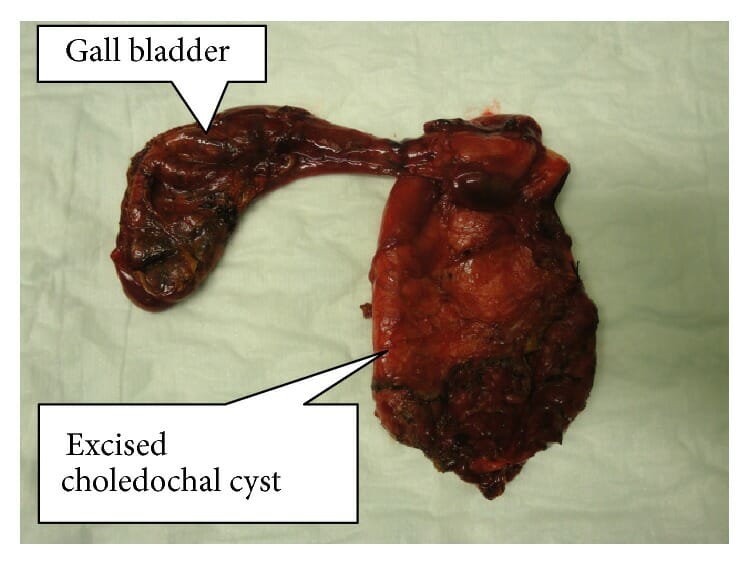
Resected specimen of completely excised choledochal cyst with the gallbladder
Image: “Resected specimen of completely excised choledochal cyst with gall bladder” by Norman Oneil Machado et al. License: CC BY 4.0