


A colestase em recém-nascidos e pequenos lactentes corresponde a uma hiperbilirrubinemia conjugada nos 1ºs 3 meses de vida decorrente de uma excreção biliar alterada. As malformações da via biliar que envolvem a vesícula e o ducto biliares são agrupadas em colangiopatias obliterativas quísticas e não quísticas, sendo a mais comum a atresia biliar. Causas menos comuns incluem a síndrome genética de Alagille, causas infecciosas e distúrbios metabólicos. A apresentação clínica é com icterícia obstrutiva. A ecografia e a CPRM são ferramentas de diagnóstico úteis, e, por vezes, é possível realizar um diagnóstico pré-natal através da ecografia. As causas quísticas requerem frequentemente cirurgia para corrigir o defeito e permitir o crescimento normal da criança. Pode ser necessário um transplante hepático em casos de atresia biliar com hipertensão portal.
Last updated: Feb 16, 2026
A colestase em recém-nascidos e pequenos lactentes corresponde a uma hiperbilirrubinemia conjugada nos 1ºs 3 meses de vida decorrente de uma excreção biliar alterada.
As malformações da via biliar que envolvem a vesícula e o ducto biliares são agrupadas em colangiopatias obliterativas quísticas e não quísticas, sendo a mais MAIS Androgen Insensitivity Syndrome comum a atresia Atresia Hypoplastic Left Heart Syndrome (HLHS) biliar. Causas menos comuns incluem a síndrome genética de Alagille, causas infecciosas e distúrbios metabólicos.
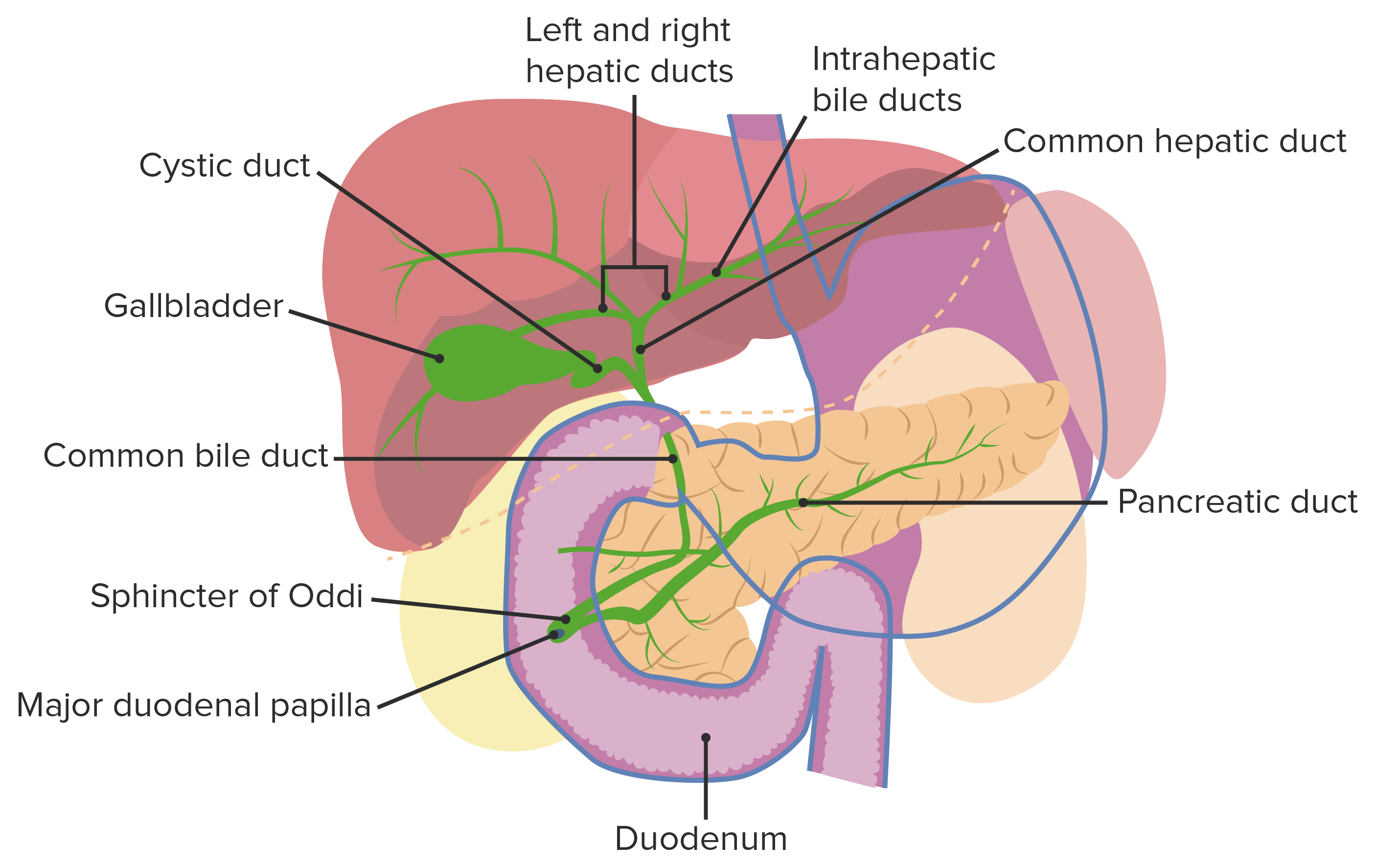
Anatomia normal da via biliar
Imagem por Lecturio.A atresia Atresia Hypoplastic Left Heart Syndrome (HLHS) biliar é uma obliteração idiopática, progressiva e fibrótica da árvore biliar extra-hepática, cuja apresentação é uma obstrução biliar nos primeiros 3 meses de vida.
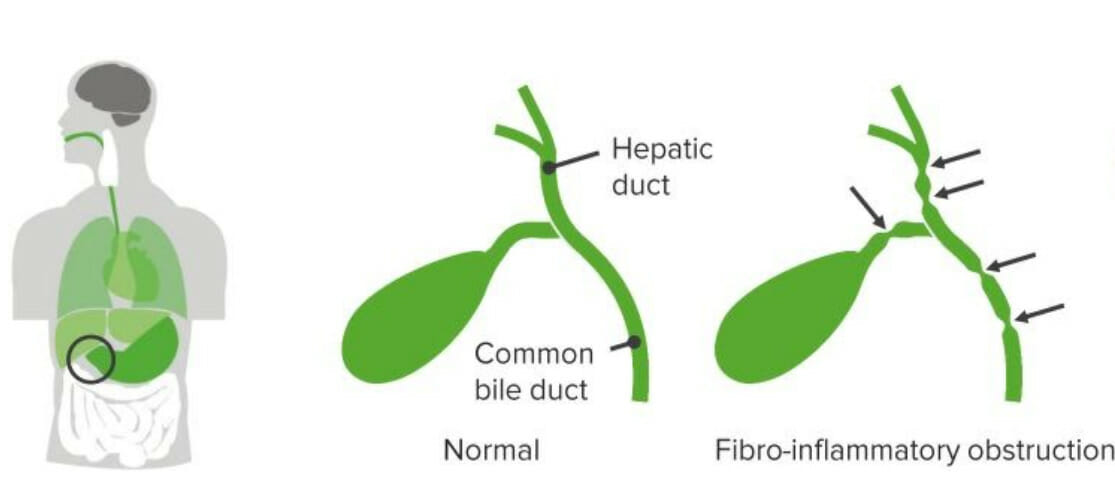
Atresia biliar
Imagem por Lecturio.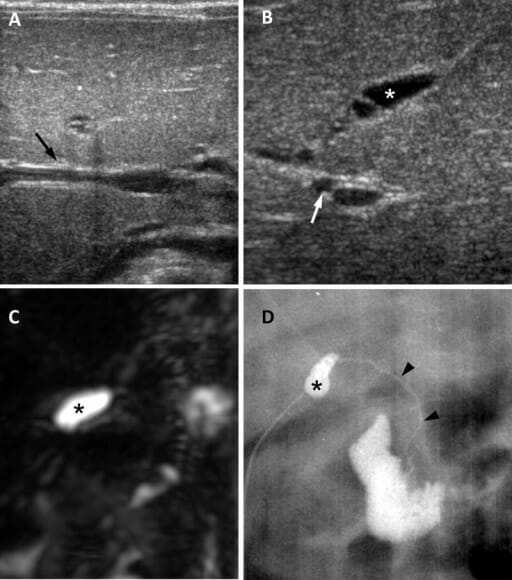
Atresia biliar numa rapariga com 65 dias:
A: Nesta ecografia, no plano transversal, o sinal do cordão triangular é negativo (seta).B: A imagem ecográfica no plano subcostal oblíquo mostra uma atresia da vesícula biliar, que mede 0,8 cm (asterisco), e uma artéria hepática aumentada com 1,5 mm (seta).
C: Imagem tridimensional de CPRM com uma árvore biliar extra-hepática não visível e uma vesícula biliar pequena (asterisco).
D: Imagem de colangiografia cirúrgica onde se vê uma vesícula biliar pequena (asterisco) com um ducto biliar comum patente, mas extremamente hipoplásico (cabeças de seta).
Imagem : “Biliary atresia in a 65-day-old girl” pelo Department of Radiology and Center for Imaging Science, Samsung Medical Center, Sungkyunkwan University School of Medicine. Licença: CC BY 4.0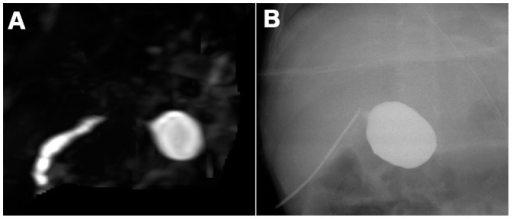
A: Atresia biliar num lactente de 90 dias. Na CPRM tridimensional não há exibição dos ductos biliares extra-hepáticos, exceto pelo ducto biliar comum cístico.
B: Colangiografia intraoperatória do mesmo lactente.
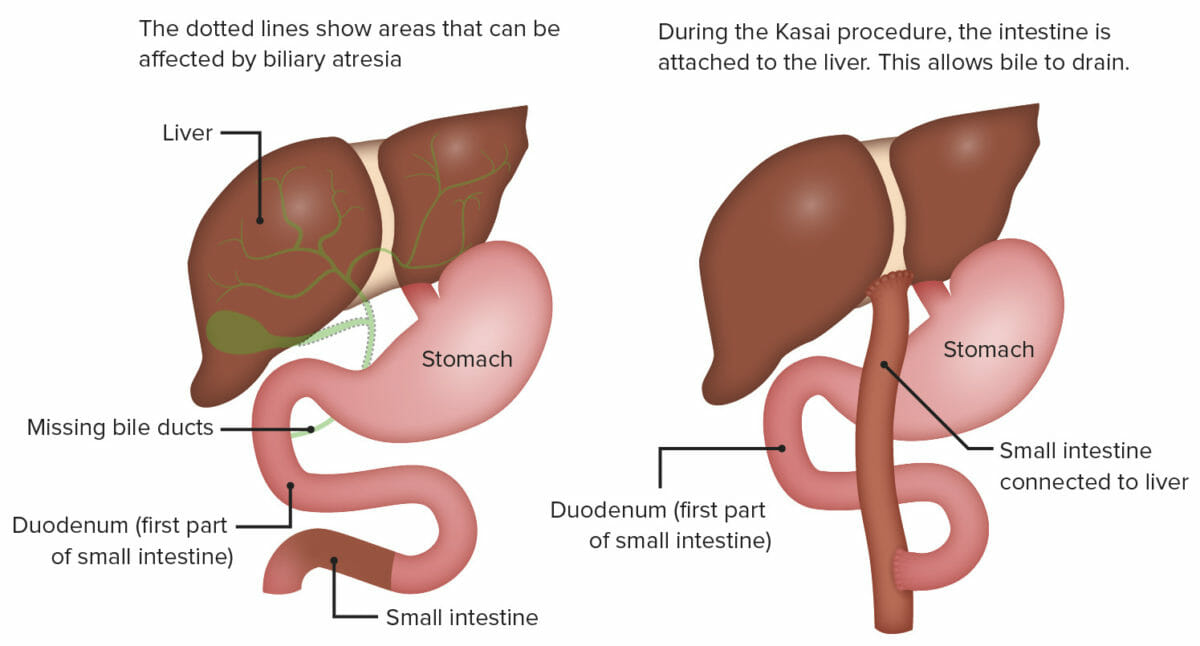
Porto-enterostomia (procedimento de Kasai):
Nesta cirurgia há exposição da porta hepatis, através de uma excisão radical de todo o tecido ductal biliar até a cápsula hepática, e criação de uma anastomose em Y de Roux com uma alça do jejuno na cápsula hepática exposta, acima da bifurcação da veia porta.
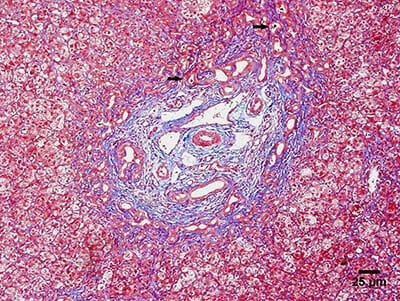
Fotomicrografia onde se observam os principais achados histológicos da atresia biliar:
Biópsia hepática com proliferação ductular e rolhões biliares (setas).
A síndrome de Alagille é uma doença genética caracterizada por uma escassez de ductos biliares no fígado, o que culmina em colestase. Esta síndrome afeta também outros órgãos.
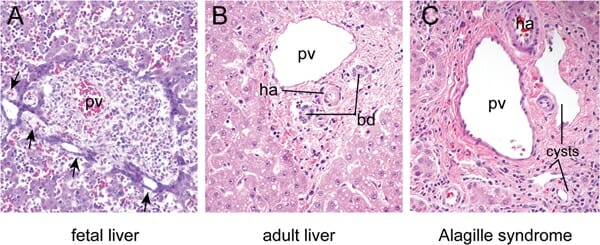
A: Fígado fetal às 16 semanas de desenvolvimento, onde se observam dilatações focais (setas) nos precursores das células epiteliais biliares em redor da veia porta (PV, pela sigla em inglês).
B: Fígado maduro com os ductos biliares (BD, pela sigla em inglês) e artéria hepática (HA, pela sigla em inglês) embutidos no mesênquima periportal num arranjo característico, conhecido como a “tríade portal”. Os hepatócitos dispostos em cordas circundam esta tríade.
C: Região da tríade portal de um indivíduo com síndrome de Alagille, com quistos ductais em vez dos ductos biliares normais, devido a defeitos na remodelação da placa ductal.
Os quistos do colédoco são anomalias congénitas do ducto biliar com um aumento anormal do tamanho e obstrução dos ductos biliares intra e/ou extra-hepáticos.
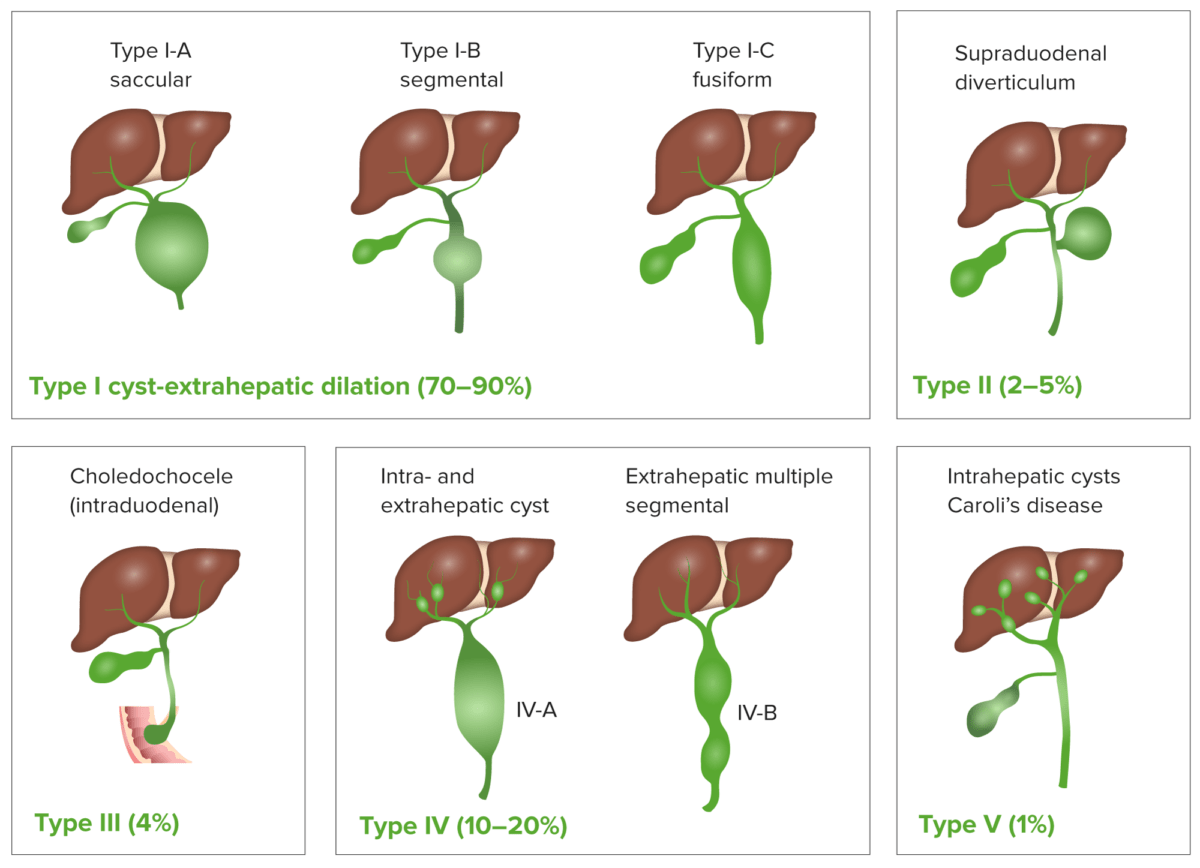
Classificação dos quistos do colédoco
Imagem por Lecturio.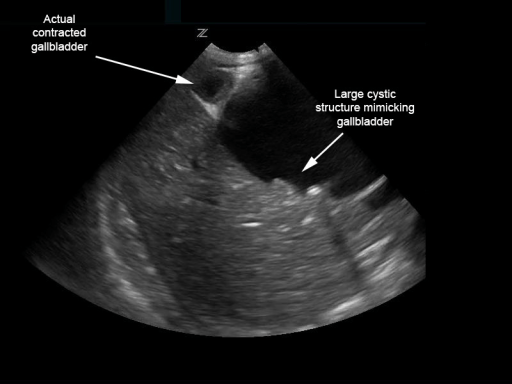
No entanto, a vesícula biliar contraída encontra-se adjacente.
Imagem : “Biliary choledochal cyst” pelo University of Massachusetts, Department of Emergency Medicine, Boston. Licença: CC BY 4.0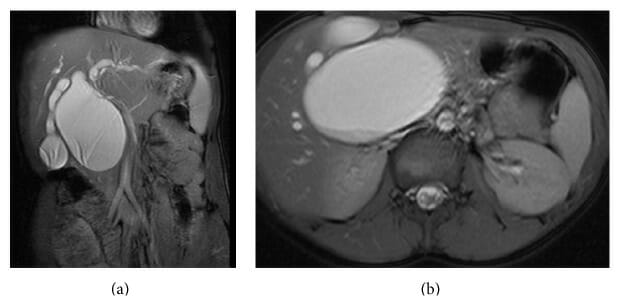
(a): CPRM com um quisto sacular do colédoco tipo IA.
(b): CPRM (corte transversal) com um quisto sacular do colédoco tipo IA grande.
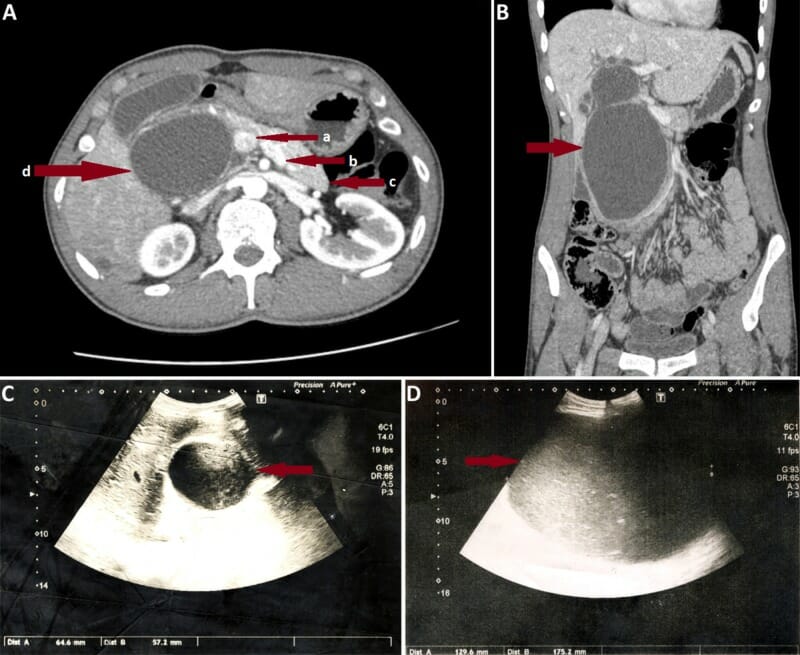
A: Incidência coronal de TC abdominal num indivíduo com um quisto do colédoco tipo I grande. As setas vermelhas apontam a veia porta (a), veia esplénica (b), corpo e cauda do pâncreas (c), e quisto do colédoco gigante (d), que oblitera a cabeça pancreática.
B: TC axial do mesmo indivíduo.
C: Imagem ecográfica do mesmo indivíduo no dia do internamento. É de salientar o rápido crescimento do quisto, observado no 7º dia de internamento (D).
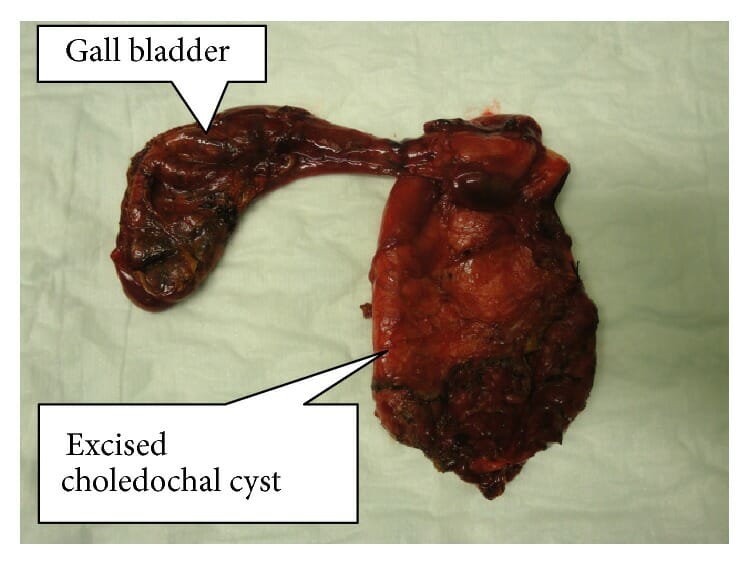
Material da resseção de um quisto do colédoco completamente excisado juntamente com a vesícula biliar
Imagem : “Resected specimen of completely excised choledochal cyst with gall bladder” pelo Norman Oneil Machado et al. Licença: CC BY 4.0