Metachromatic leukodystrophy (MLD) is an inherited lysosomal storage disorder that affects myelin in the brain Brain The part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem. Nervous System: Anatomy, Structure, and Classification and spinal cord Spinal cord The spinal cord is the major conduction pathway connecting the brain to the body; it is part of the CNS. In cross section, the spinal cord is divided into an H-shaped area of gray matter (consisting of synapsing neuronal cell bodies) and a surrounding area of white matter (consisting of ascending and descending tracts of myelinated axons). Spinal Cord: Anatomy. Genetic mutations Genetic Mutations Carcinogenesis result in the creation of a dysfunctional arylsulfatase A (ARSA) enzyme, which is unable to break down cerebroside Cerebroside Neutral glycosphingolipids that contain a monosaccharide, normally glucose or galactose, in 1-ortho-beta-glycosidic linkage with the primary alcohol of an n-acyl sphingoid (ceramide). In plants the monosaccharide is normally glucose and the sphingoid usually phytosphingosine. In animals, the monosaccharide is usually galactose, though this may vary with the tissue and the sphingoid is usually sphingosine or dihydrosphingosine. Fatty Acids and Lipids sulfate. The accumulation of this metabolite results in permanent damage to oligodendroglial and Schwann cells (myelin) in both the central and peripheral nervous systems. Patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship can present with seizures Seizures A seizure is abnormal electrical activity of the neurons in the cerebral cortex that can manifest in numerous ways depending on the region of the brain affected. Seizures consist of a sudden imbalance that occurs between the excitatory and inhibitory signals in cortical neurons, creating a net excitation. The 2 major classes of seizures are focal and generalized. Seizures, weakness, and behavioral and personality changes. Blood enzyme and urine testing for ARSA-A and sulfatides Sulfatides Mycobacterium, respectively, are the best diagnostic tests Diagnostic tests Diagnostic tests are important aspects in making a diagnosis. Some of the most important epidemiological values of diagnostic tests include sensitivity and specificity, false positives and false negatives, positive and negative predictive values, likelihood ratios, and pre-test and post-test probabilities. Epidemiological Values of Diagnostic Tests. Treatment is challenging and usually focuses on relieving symptoms, although new gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics therapies are available for specific subgroups of MLD patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship. Serious complications include dementia Dementia Major neurocognitive disorders (NCD), also known as dementia, are a group of diseases characterized by decline in a person's memory and executive function. These disorders are progressive and persistent diseases that are the leading cause of disability among elderly people worldwide. Major Neurocognitive Disorders and seizures Seizures A seizure is abnormal electrical activity of the neurons in the cerebral cortex that can manifest in numerous ways depending on the region of the brain affected. Seizures consist of a sudden imbalance that occurs between the excitatory and inhibitory signals in cortical neurons, creating a net excitation. The 2 major classes of seizures are focal and generalized. Seizures.
Last updated: Apr 23, 2025
Metachromatic leukodystrophy (MLD) is an autosomal recessive Autosomal recessive Autosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal recessive diseases are only expressed when 2 copies of the recessive allele are inherited. Autosomal Recessive and Autosomal Dominant Inheritance white matter White Matter The region of central nervous system that appears lighter in color than the other type, gray matter. It mainly consists of myelinated nerve fibers and contains few neuronal cell bodies or dendrites. Brown-Séquard Syndrome disease caused by a deficiency in arylsulfatase A resulting in accumulation of cerebroside Cerebroside Neutral glycosphingolipids that contain a monosaccharide, normally glucose or galactose, in 1-ortho-beta-glycosidic linkage with the primary alcohol of an n-acyl sphingoid (ceramide). In plants the monosaccharide is normally glucose and the sphingoid usually phytosphingosine. In animals, the monosaccharide is usually galactose, though this may vary with the tissue and the sphingoid is usually sphingosine or dihydrosphingosine. Fatty Acids and Lipids sulfate. This accumulation leads to the destruction of myelin and causes neurologic deficits Neurologic Deficits High-Risk Headaches.
Based on age at symptom onset:
Consider diagnostic testing in patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship expressing typical clinical features.
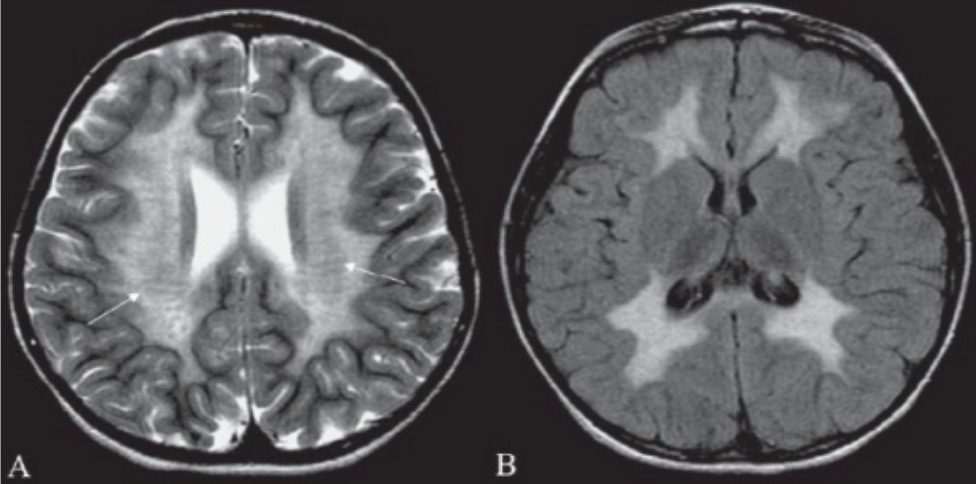
Brain MRI of a patient with metachromatic leukodystrophy:
Brain MRI showing hyperintensity in periventricular white matter (A) and in subcortical supratentorial white matter (B) can help confirm laboratory diagnosis of metachromatic leukodystrophy.
No curative therapy for MLD is widely available. Umbilical cord Umbilical cord The flexible rope-like structure that connects a developing fetus to the placenta in mammals. The cord contains blood vessels which carry oxygen and nutrients from the mother to the fetus and waste products away from the fetus. Placenta, Umbilical Cord, and Amniotic Cavity blood transplantation for children with presymptomatic late-infantile MLD or minimally symptomatic juvenile MLD can slow disease progression.
Supportive therapy is preferred for patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship with symptoms and for all patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship with juvenile and late-onset forms MLD:
Clinical trials of recombinant human arylsulfatase A (rhARSA) enzyme demonstrated its safety in children with late-infantile MLD.