


A leucodistrofia metacromática ( MLD MLD Metachromatic leukodystrophy (MLD) is an inherited lysosomal storage disorder that affects myelin in the brain and spinal cord. Genetic mutations result in the creation of a dysfunctional arylsulfatase a (arsa) enzyme, which is unable to break down cerebroside sulfate. The accumulation of this metabolite results in permanent damage to oligodendroglial and schwann cells (myelin). Metachromatic Leukodystrophy) é um distúrbio de armazenamento lisossomal hereditário que afeta a mielina no cérebro e na medula espinhal. Mutações genéticas resultam na criação de uma enzima disfuncional arilsulfatase A (ARSA), que é incapaz de quebrar o sulfato de cerebrosídeo. O acúmulo desse metabólito resulta em dano permanente às células oligodendrogliais e de Schwann (mielina) tanto no sistema nervoso central quanto no periférico. Os pacientes podem apresentar convulsões, fraqueza e alterações comportamentais e de personalidade. Enzimas sanguíneas e exames de urina para ARSA-A e sulfatos, respectivamente, são os melhores testes Testes Gonadal Hormones diagnósticos. O tratamento é desafiador e geralmente se concentra no alívio dos sintomas, embora novas terapias genéticas estejam disponíveis para subgrupos específicos de pacientes com DLM. As complicações graves incluem demência e convulsões.
Last updated: Apr 23, 2025
A leucodistrofia metacromática ( MLD MLD Metachromatic leukodystrophy (MLD) is an inherited lysosomal storage disorder that affects myelin in the brain and spinal cord. Genetic mutations result in the creation of a dysfunctional arylsulfatase a (arsa) enzyme, which is unable to break down cerebroside sulfate. The accumulation of this metabolite results in permanent damage to oligodendroglial and schwann cells (myelin). Metachromatic Leukodystrophy) é uma doença autossômica recessiva da substância branca causada por uma deficiência na arilsulfatase A, resultando no acúmulo de sulfato de cerebrosídeo. Esse acúmulo leva à destruição da mielina e causa déficits neurológicos.
Com base na idade de início dos sintomas:
Considere testes Testes Gonadal Hormones diagnósticos em pacientes que expressam características clínicas típicas.
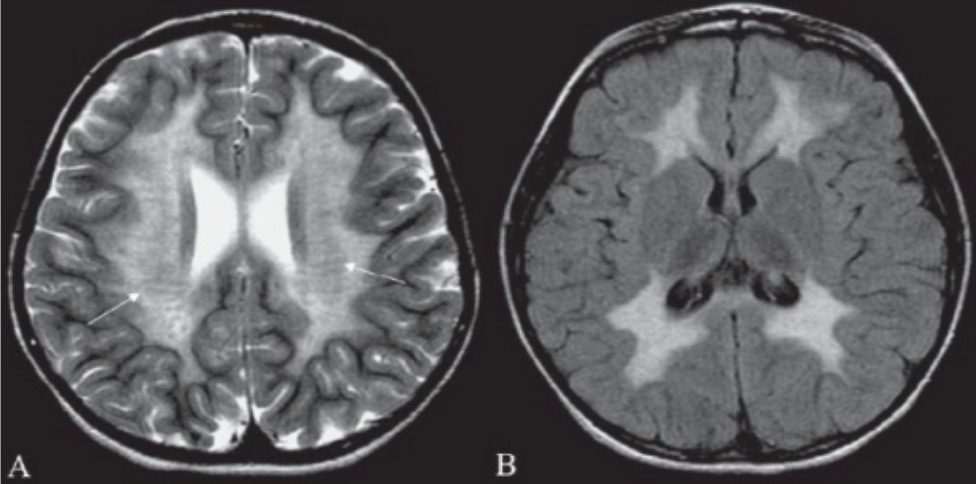
RM cerebral de um paciente com leucodistrofia metacromática:
RM cerebral mostrando hiperintensidade na substância branca periventricular (A) e na substância branca supratentorial subcortical (B) pode ajudar a confirmar o diagnóstico laboratorial de leucodistrofia metacromática.
Nenhuma terapia curativa para MLD MLD Metachromatic leukodystrophy (MLD) is an inherited lysosomal storage disorder that affects myelin in the brain and spinal cord. Genetic mutations result in the creation of a dysfunctional arylsulfatase a (arsa) enzyme, which is unable to break down cerebroside sulfate. The accumulation of this metabolite results in permanent damage to oligodendroglial and schwann cells (myelin). Metachromatic Leukodystrophy está amplamente disponível. O transplante de sangue do cordão umbilical para crianças com DLM pré-sintomática infantil tardia ou DLM juvenil minimamente sintomática pode retardar a progressão da doença.
A terapia de suporte é preferida para pacientes com sintomas e para todos os pacientes com formas juvenil e de início tardio de MLD MLD Metachromatic leukodystrophy (MLD) is an inherited lysosomal storage disorder that affects myelin in the brain and spinal cord. Genetic mutations result in the creation of a dysfunctional arylsulfatase a (arsa) enzyme, which is unable to break down cerebroside sulfate. The accumulation of this metabolite results in permanent damage to oligodendroglial and schwann cells (myelin). Metachromatic Leukodystrophy:
Ensaios clínicos da enzima arilsulfatase A humana recombinante ( rhARSA rhARSA Metachromatic Leukodystrophy) demonstraram sua segurança em crianças com DLM infantil tardia.
