5-alpha-reductase deficiency is an autosomal recessive Autosomal recessive Autosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal recessive diseases are only expressed when 2 copies of the recessive allele are inherited. Autosomal Recessive and Autosomal Dominant Inheritance intersex or “disorder of sex Sex The totality of characteristics of reproductive structure, functions, phenotype, and genotype, differentiating the male from the female organism. Gender Dysphoria development” (DSD) condition caused by a loss-of-function mutation Mutation Genetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations in a gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics on chromosome Chromosome In a prokaryotic cell or in the nucleus of a eukaryotic cell, a structure consisting of or containing DNA which carries the genetic information essential to the cell. Basic Terms of Genetics 2. The affected subjects have a 46,XY karyotype Karyotype The full set of chromosomes presented as a systematized array of metaphase chromosomes from a photomicrograph of a single cell nucleus arranged in pairs in descending order of size and according to the position of the centromere. Congenital Malformations of the Female Reproductive System, bilateral testes Testes Gonadal Hormones, and normal testosterone Testosterone A potent androgenic steroid and major product secreted by the leydig cells of the testis. Its production is stimulated by luteinizing hormone from the pituitary gland. In turn, testosterone exerts feedback control of the pituitary LH and FSH secretion. Depending on the tissues, testosterone can be further converted to dihydrotestosterone or estradiol. Androgens and Antiandrogens production but have impaired virilization during embryogenesis due to defective conversion of testosterone Testosterone A potent androgenic steroid and major product secreted by the leydig cells of the testis. Its production is stimulated by luteinizing hormone from the pituitary gland. In turn, testosterone exerts feedback control of the pituitary LH and FSH secretion. Depending on the tissues, testosterone can be further converted to dihydrotestosterone or estradiol. Androgens and Antiandrogens to dihydrotestosterone Dihydrotestosterone A potent androgenic metabolite of testosterone. It is produced by the action of the enzyme 3-oxo-5-alpha-steroid 4-dehydrogenase. Gonadal Hormones ( DHT DHT A potent androgenic metabolite of testosterone. It is produced by the action of the enzyme 3-oxo-5-alpha-steroid 4-dehydrogenase. Gonadal Hormones), which is a significantly more potent androgen. This leads to male pseudohermaphroditism or ambiguous genitalia Ambiguous Genitalia Primary Amenorrhea in males. Also known as pseudovaginal perineoscrotal hypospadias Hypospadias A birth defect due to malformation of the urethra in which the urethral opening is below its normal location. In the male, the malformed urethra generally opens on the ventral surface of the penis or on the perineum. In the female, the malformed urethral opening is in the vagina. Penile Anomalies and Conditions, these patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship present with a clitoris-like phallus, cryptorchidism Cryptorchidism Cryptorchidism is one of the most common congenital anomalies in young boys. Typically, this asymptomatic condition presents during a routine well-child examination where 1 or both testicles are not palpable in the scrotum. Cryptorchidism, bifid scrotum Scrotum A cutaneous pouch of skin containing the testicles and spermatic cords. Testicles: Anatomy, and a rudimentary prostate Prostate The prostate is a gland in the male reproductive system. The gland surrounds the bladder neck and a portion of the urethra. The prostate is an exocrine gland that produces a weakly acidic secretion, which accounts for roughly 20% of the seminal fluid. . No Müllerian structures Müllerian structures Congenital Disorders of Sexual Development are present.
Last updated: Dec 15, 2025
Synonyms
Epidemiology
Etiology
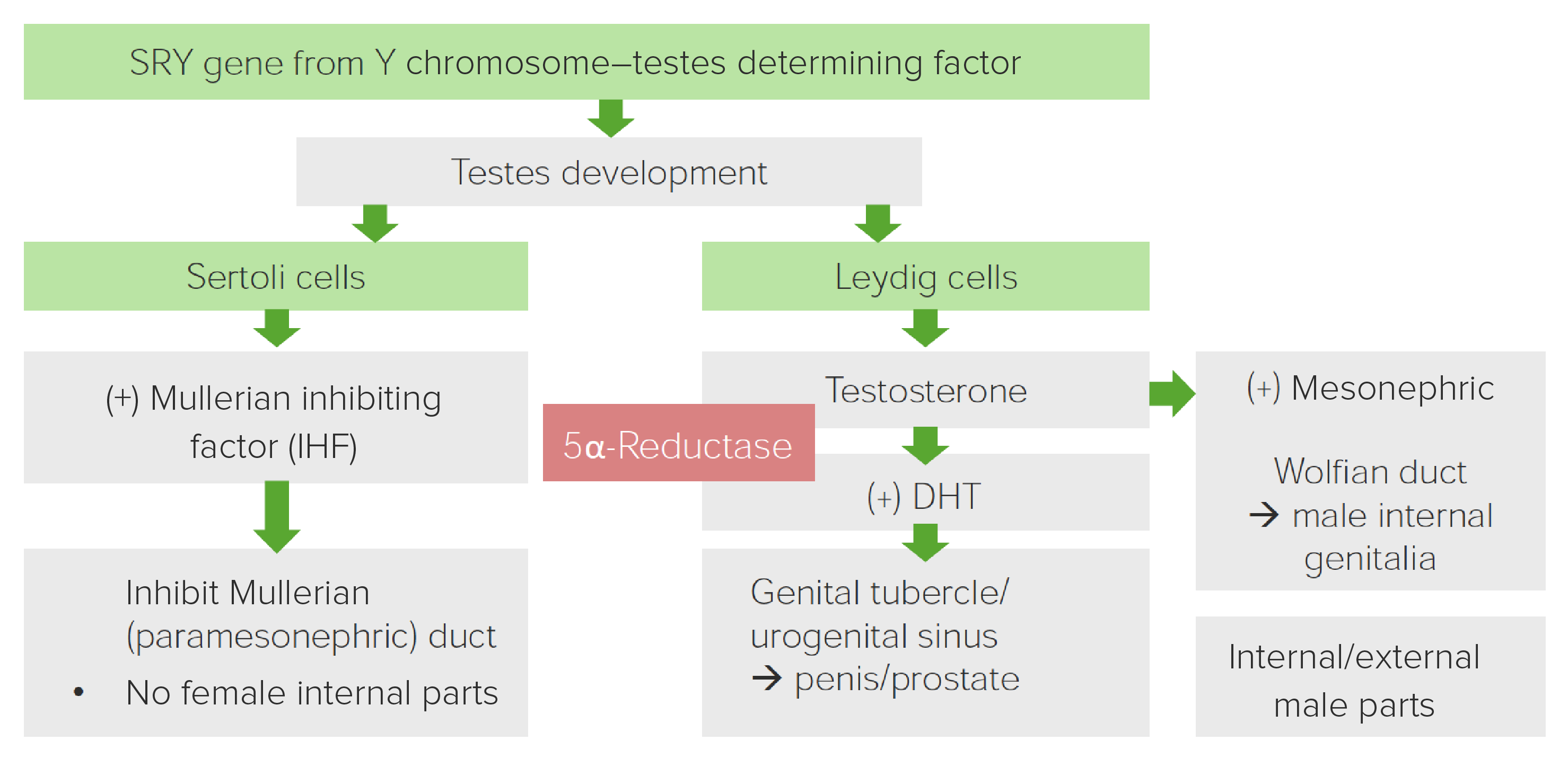
Diagram displaying the role of 5α-Reductase in the development of male genitalia
Image by Lecturio.| Clinical phenotype Phenotype The complete genetic complement contained in the DNA of a set of chromosomes in a human. The length of the human genome is about 3 billion base pairs. Basic Terms of Genetics |
|
|---|---|
| Cognition/intelligence | Normal |
| Behavioral phenotype Phenotype The complete genetic complement contained in the DNA of a set of chromosomes in a human. The length of the human genome is about 3 billion base pairs. Basic Terms of Genetics | Normal |
| Associations | Normal internal male urogenital organs |
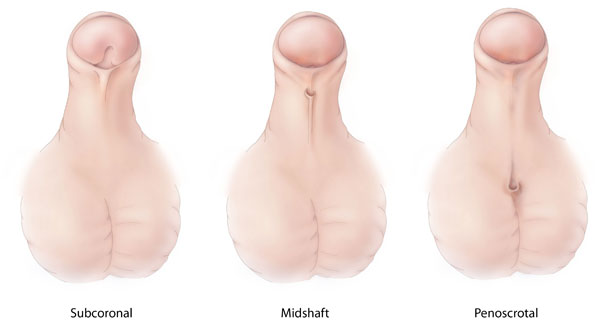
Different types of hypospadias, a congenital malformation in which the opening of the penile urethra is abnormally located on the ventral side of the penis, scrotum, or perineum.
Image: “Hypospadias-lg” by Centers for Disease Control and Prevention. License: CC0 1.0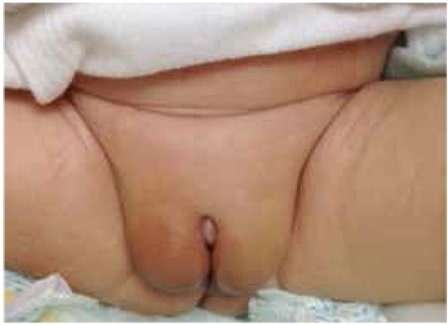
Genitalia of male neonate with 5-alpha-reductase deficiency presenting with apparently normal external female genitalia with bilaterally palpable gonads in the labial folds. Length of the phallus/clitoris was around 0.5 cm.
Image: “ Genitalia of index case at birth” by U.S. National Library of Medicine. License: CC BY 2.5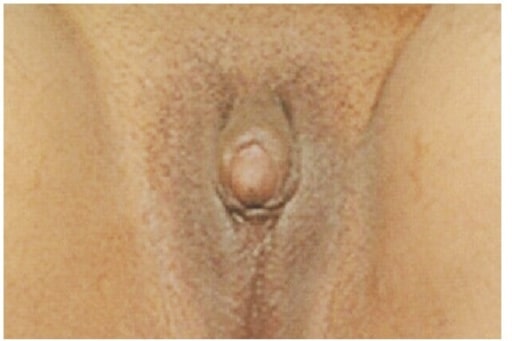
Clitoromegaly and ambiguous genitalia on a 21-year-old complaining of primary amenorrhea and lack of breast development. Physical examination revealed that gonads were palpable in the inguinal canal bilaterally, gynaecological examination showed a 5 cm blind vaginal pouch, and ultrasonography of abdomen and pelvis showed atrophic uterus and bilateral ill-defined gonad-like structures. A chromosomal study revealed a 46,XY karyotype.
Image: “Clitoromegaly and ambiguous genitalia” by Department of Internal Medicine, Imam Khomeini Hospital, Ardabil University of Medical Sciences, Ardabil, Iran. License: CC BY 3.0The following conditions are differential diagnoses for 5-alpha-reductase deficiency:
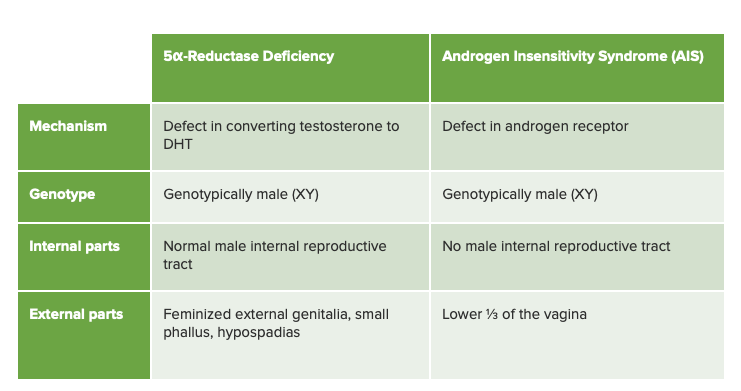
Chart comparing 5α-reductase deficiency and androgen insensitivity syndrome, its closest differential diagnosis
Image by Lecturio.