La deficiencia de 5-alfa-reductasa es una condición intersexual autosómica recesiva o un «trastorno del desarrollo sexual» (TDS) causada por una mutación de pérdida de función en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum un gen del cromosoma 2. Los LOS Neisseria sujetos afectados tienen un cariotipo 46,XY, testículos bilaterales y una producción normal de testosterona, pero tienen una virilización alterada durante la embriogénesis debido a la conversión defectuosa de testosterona en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum dihidrotestosterona ( DHT DHT A potent androgenic metabolite of testosterone. It is produced by the action of the enzyme 3-oxo-5-alpha-steroid 4-dehydrogenase. Gonadal Hormones), que es un andrógeno significativamente más potente. Esto da lugar a un pseudohermafroditismo masculino o a genitales ambiguos en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum los LOS Neisseria varones. También conocido como hipospadias perineoescrotal pseudovaginal, estos pacientes presentan un falo similar al AL Amyloidosis clítoris, criptorquidia, escroto bífido y una próstata rudimentaria. No hay estructuras müllerianas presentes.
Last updated: Dec 15, 2025
Sinónimos
Epidemiología
Etiología
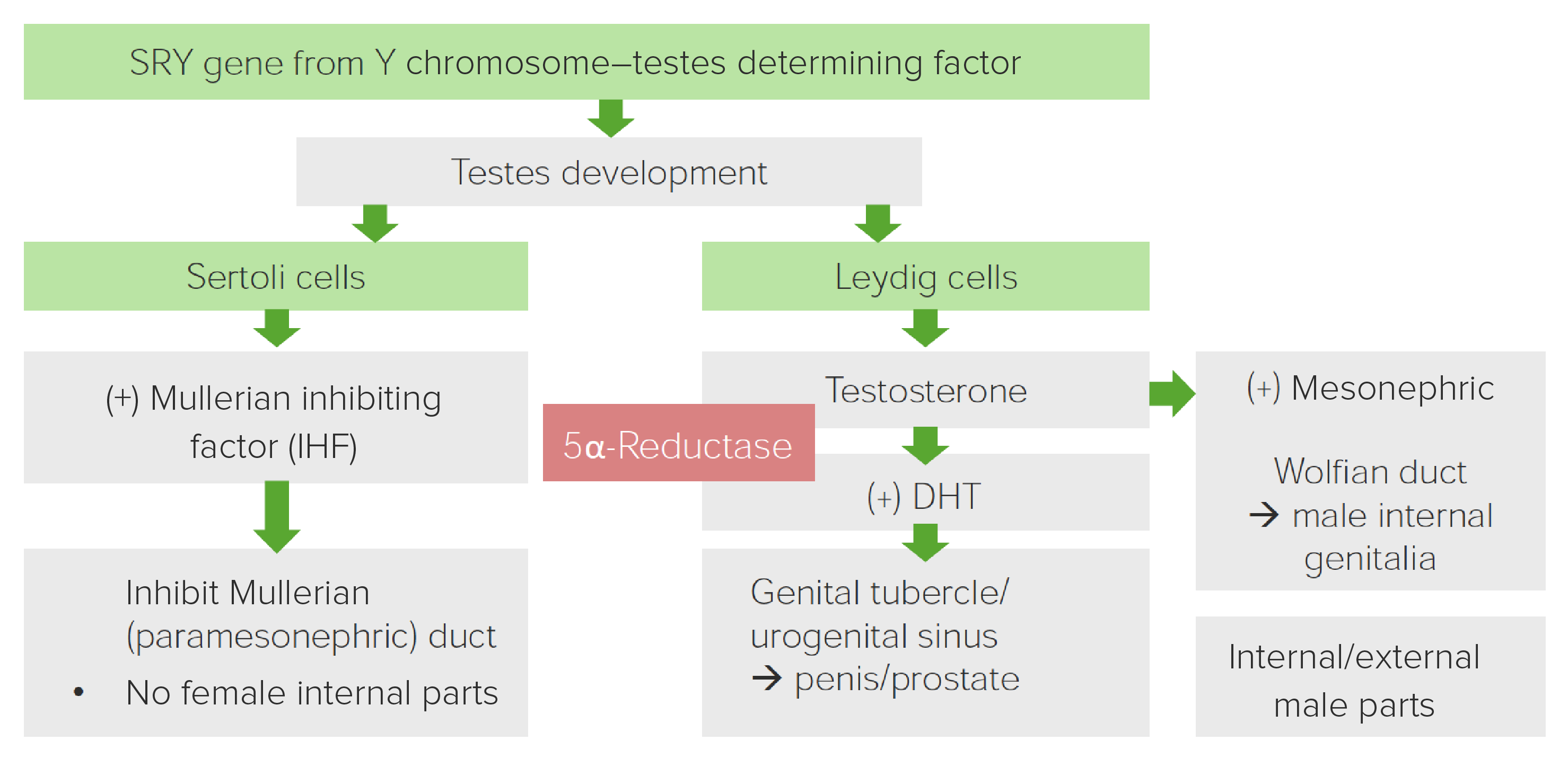
Diagrama que muestra el papel de la 5-alfa-reductasa en el desarrollo de los genitales masculinos
Imagen por Lecturio.| Fenotipo clínico |
|
|---|---|
| Cognición/inteligencia | Normal |
| Fenotipo conductual | Normal |
| Asociaciones | Órganos urogenitales masculinos internos normales |
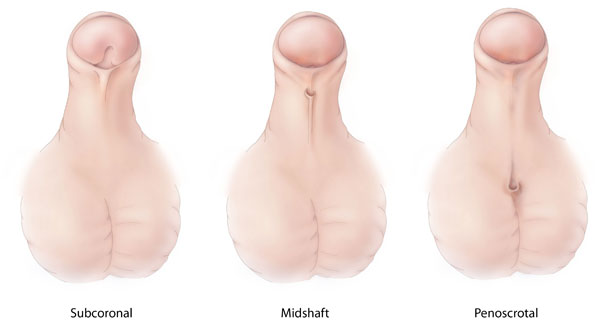
Diferentes tipos de hipospadias, una malformación congénita en la que el orificio de la uretra del pene está anormalmente situado en la parte ventral del pene, el escroto o el periné.
Imagen: “Hypospadias-lg” by Centers for Disease Control and Prevention. Licencia: CC0 1.0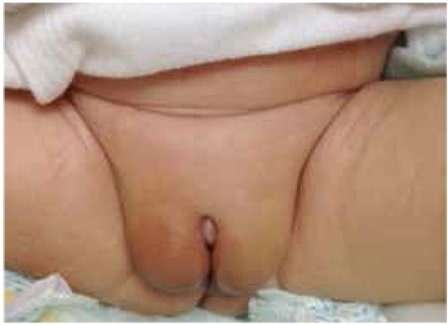
Genitales de un neonato masculino con deficiencia de 5-alfa-reductasa que presenta genitales femeninos externos aparentemente normales con gónadas palpables bilateralmente en los pliegues labiales. La longitud del falo/clítoris era de unos 0,5 cm.
Imagen: “Genitalia of index case at birth” by U.S. National Library of Medicine. Licencia: CC BY 2.5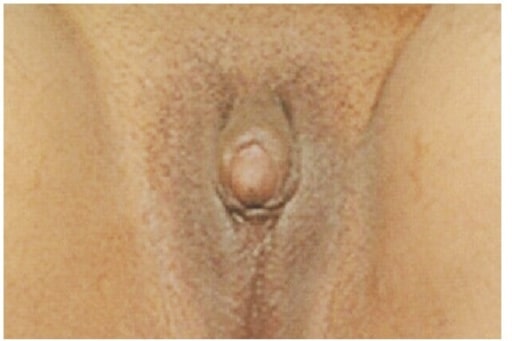
Clitoromegalia y genitales ambiguos en una joven de 21 años que se queja de amenorrea primaria y falta de desarrollo mamario. El examen físico reveló que las gónadas eran palpables en el canal inguinal bilateralmente, el examen ginecológico mostró una bolsa vaginal ciega de 5 cm, y la ecografía de abdomen y pelvis mostró un útero atrófico y estructuras bilaterales mal definidas similares a las gónadas. Un estudio cromosómico reveló un cariotipo 46,XY.
Imagen: “Clitoromegaly and ambiguous genitalia” by Department of Internal Medicine, Imam Khomeini Hospital, Ardabil University of Medical Sciences, Ardabil, Iran. Licencia: CC BY 3.0Las siguientes condiciones son diagnósticos diferenciales para la deficiencia de 5-alfa-reductasa:
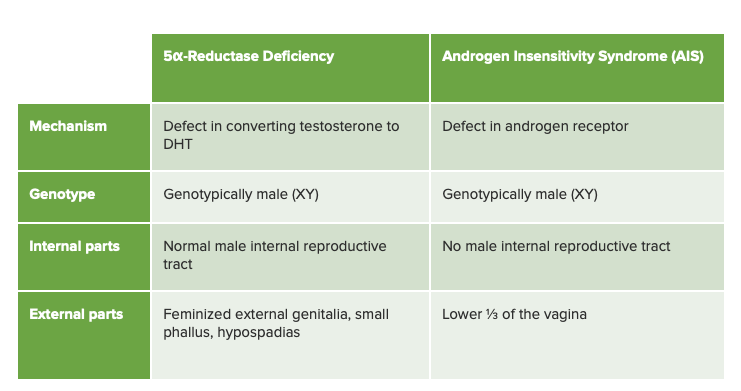
Cuadro comparativo entre la deficiencia de 5-alfa-reductasa y el síndrome de insensibilidad a los andrógenos, su diagnóstico diferencial más cercano
Imagen por Lecturio.