


A deficiência de 5-alfa-redutase é uma doença intersexual autossómica recessiva ou “perturbação do desenvolvimento sexual” (PDS) causada por uma mutação de perda de função num gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics no cromossoma 2. Os indivíduos afetados têm um cariótipo 46,XY, testículos bilaterais e produção normal de testosterona, mas têm compromisso da virilização durante a embriogénese devido aos defeitos na conversão de testosterona em dihidrotestosterona ( DHT DHT A potent androgenic metabolite of testosterone. It is produced by the action of the enzyme 3-oxo-5-alpha-steroid 4-dehydrogenase. Gonadal Hormones), que é um androgénio significativamente mais MAIS Androgen Insensitivity Syndrome potente. Isto leva ao pseudo-hermafroditismo masculino ou genitália ambígua nos homens. Também conhecida como hipospádia perineoescrotal pseudovaginal, estes pacientes apresentam um falo semelhante ao clitóris, criptorquidia, escroto bífido e próstata rudimentar. Não está presente nenhuma estrutura Mülleriana.
Last updated: Dec 15, 2025
Sinónimos
Epidemiologia
Etiologia
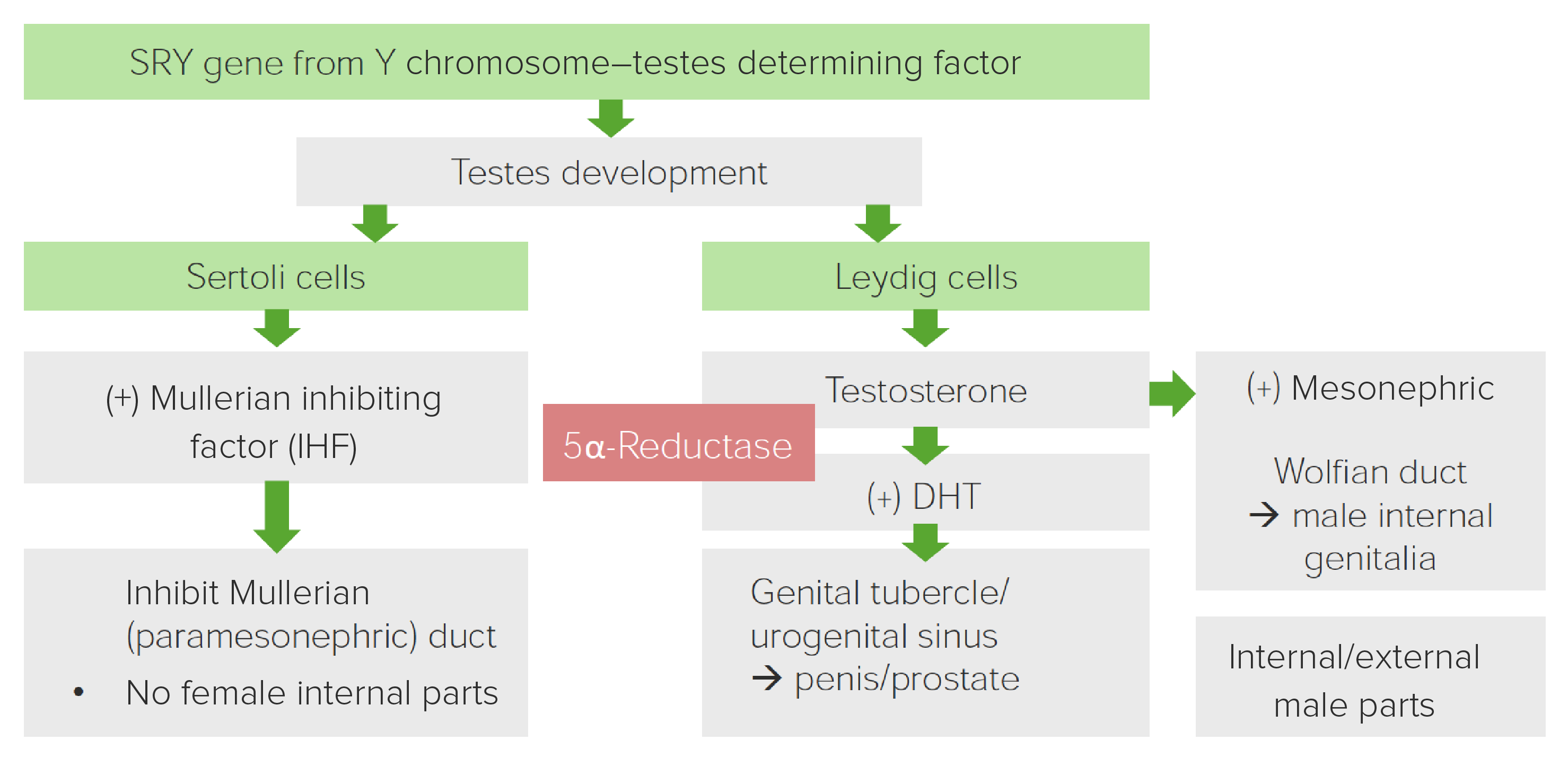
Diagrama exibindo o papel da 5α-redutase no desenvolvimento da genitália masculina
Imagem por Lecturio.| Fenótipo clínico |
|
|---|---|
| Cognição/inteligência | Normal |
| Fenótipo comportamental | Normal |
| Associações | Órgãos urogenitais internos masculinos normais |
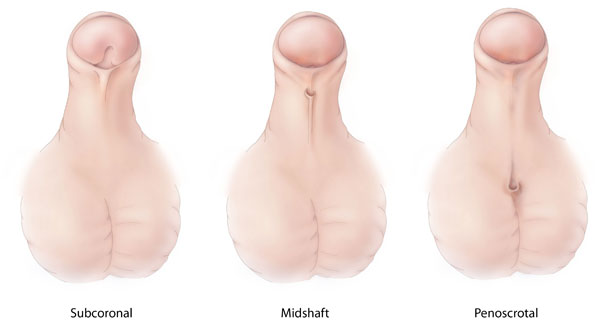
Diferentes tipos de hipospádia, uma malformação congénita em que a abertura da uretra está anormalmente localizada no lado ventral do pénis, do escroto ou do períneo.
Imagem: “Hypospadias-lg” por Centers for Disease Control and Prevention. Licença: CC0 1.0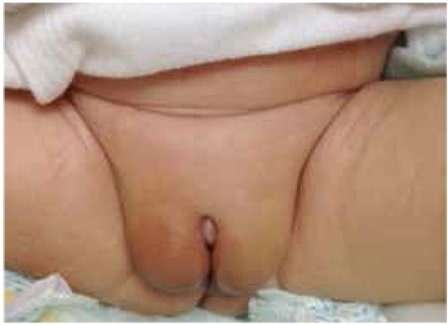
Genitália de recém-nascido do sexo masculino com deficiência de 5-alfa-redutase apresentando genitália feminina externa aparentemente normal com gónadas palpáveis bilateralmente nas pregas labiais. O comprimento do falo/clitóris era de cerca de 0,5 cm.
Imagem: “ Genitalia of index case at birth” por U.S. National Library of Medicine. Licença: CC BY 2.5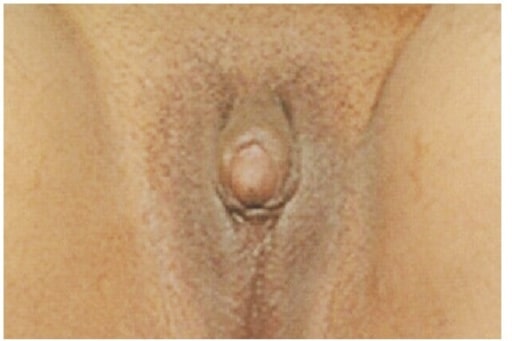
Clitoromegalia e genitália ambígua numa jovem de 21 anos com queixas de amenorreia primária e ausência de desenvolvimento mamário. O exame físico revelou que as gónadas eram palpáveis no canal inguinal bilateralmente, o exame ginecológico mostrou uma bolsa vaginal cega de 5 cm e a ecografia abdominopélvica mostrou um útero atrófico e estruturas semelhantes a gónadas bilaterais mal definidas. Um estudo cromossómico revelou um cariótipo 46, XY.
Imagem: “Clitoromegaly and ambiguous genitalia” por Department of Internal Medicine, Imam Khomeini Hospital, Ardabil University of Medical Sciences, Ardabil, Iran. Licença: CC BY 3.0As seguintes condições são diagnósticos diferenciais para a deficiência de 5-alfa-redutase:
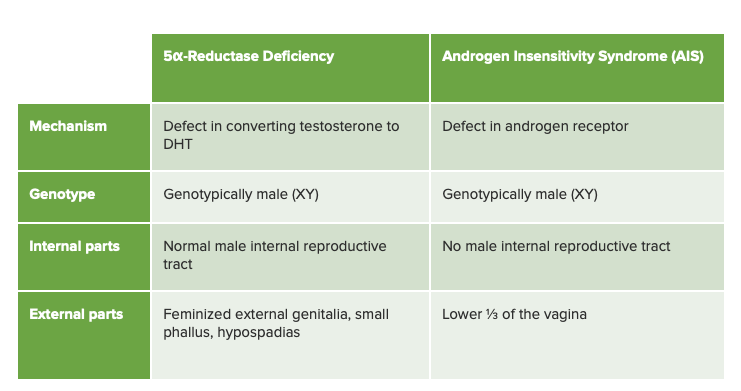
Gráfico comparando deficiência de 5α-redutase e síndrome de insensibilidade aos androgénios, o seu diagnóstico diferencial mais próximo
Imagem por Lecturio.