Interleukin-12 (IL-12) receptor Receptor Receptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell. Receptors deficiency is an autosomal recessive Autosomal recessive Autosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal recessive diseases are only expressed when 2 copies of the recessive allele are inherited. Autosomal Recessive and Autosomal Dominant Inheritance primary immunodeficiency Immunodeficiency Chédiak-Higashi Syndrome caused by a mutation Mutation Genetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations in the genes Genes A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. DNA Types and Structure that encode for the receptors Receptors Receptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell. Receptors of IL-12 on T-cells. IL-12 promotes cell-mediated immunity Cell-mediated immunity Manifestations of the immune response which are mediated by antigen-sensitized T-lymphocytes via lymphokines or direct cytotoxicity. This takes place in the absence of circulating antibody or where antibody plays a subordinate role. Squamous Cell Carcinoma (SCC) by inducing the maturation of T cells T cells Lymphocytes responsible for cell-mediated immunity. Two types have been identified - cytotoxic (t-lymphocytes, cytotoxic) and helper T-lymphocytes (t-lymphocytes, helper-inducer). They are formed when lymphocytes circulate through the thymus gland and differentiate to thymocytes. When exposed to an antigen, they divide rapidly and produce large numbers of new T cells sensitized to that antigen. T cells: Types and Functions into Th1 Th1 A subset of helper-inducer T-lymphocytes which synthesize and secrete interleukin-2; interferon-gamma; and interleukin-12. Due to their ability to kill antigen-presenting cells and their lymphokine-mediated effector activity, th1 cells are associated with vigorous delayed-type hypersensitivity reactions. T cells: Types and Functions cells and stimulating the secretion Secretion Coagulation Studies of interferon-gamma (IFN-γ). This process, in turn, activates natural killer (NK) cells, macrophages Macrophages The relatively long-lived phagocytic cell of mammalian tissues that are derived from blood monocytes. Main types are peritoneal macrophages; alveolar macrophages; histiocytes; kupffer cells of the liver; and osteoclasts. They may further differentiate within chronic inflammatory lesions to epithelioid cells or may fuse to form foreign body giant cells or langhans giant cells. Innate Immunity: Phagocytes and Antigen Presentation, and cytotoxic Cytotoxic Parvovirus B19 T cells T cells Lymphocytes responsible for cell-mediated immunity. Two types have been identified - cytotoxic (t-lymphocytes, cytotoxic) and helper T-lymphocytes (t-lymphocytes, helper-inducer). They are formed when lymphocytes circulate through the thymus gland and differentiate to thymocytes. When exposed to an antigen, they divide rapidly and produce large numbers of new T cells sensitized to that antigen. T cells: Types and Functions. IL-12 receptor Receptor Receptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell. Receptors deficiency results in impairments of all of these immunologic functions; consequently, patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship present with disseminated infections Infections Invasion of the host organism by microorganisms or their toxins or by parasites that can cause pathological conditions or diseases. Chronic Granulomatous Disease.
Last updated: Mar 28, 2025
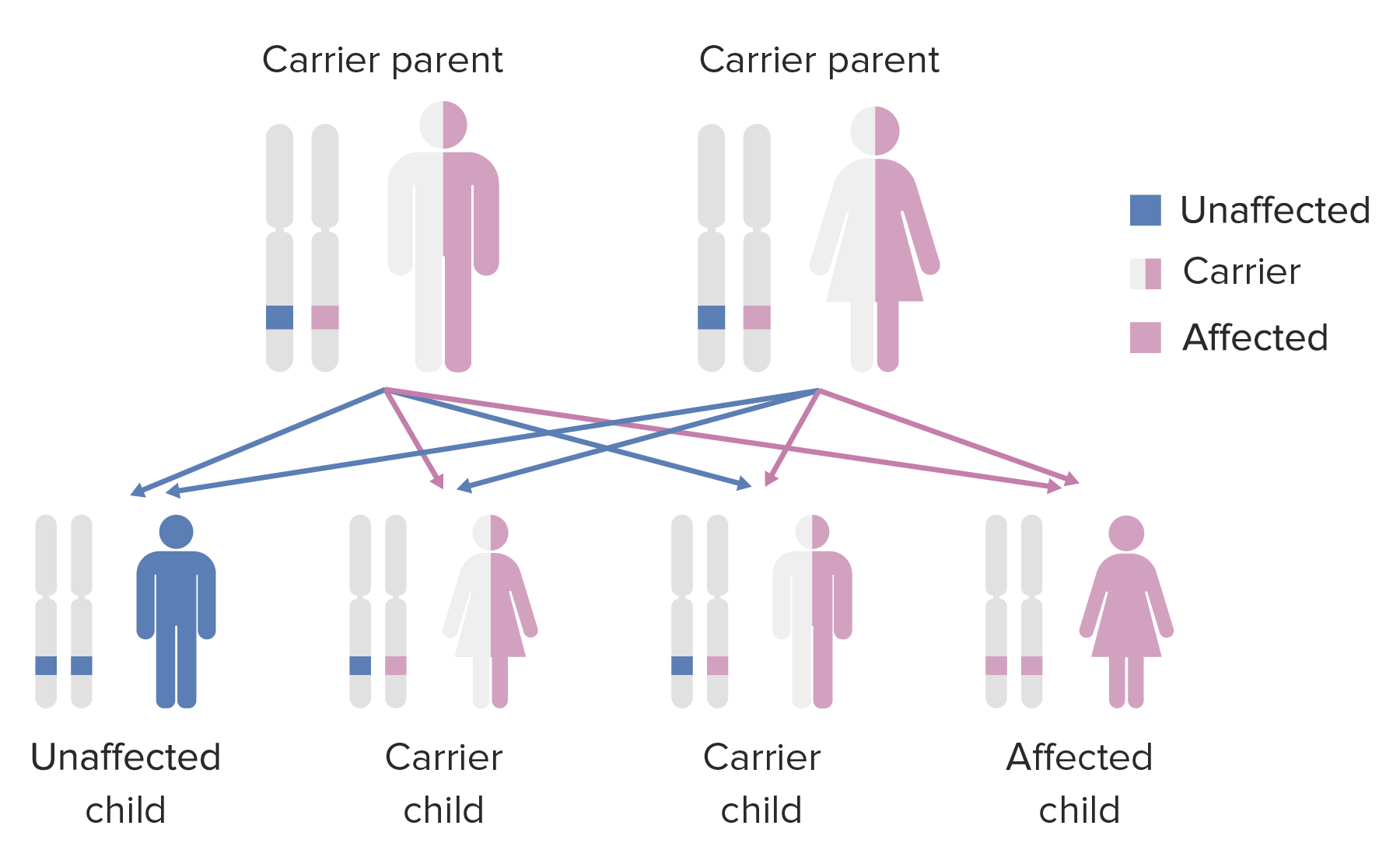
Autosomal recessive inheritance:
Affected offspring will have unaffected parents who are carriers.
Diagnosis
Management
The following conditions are differential diagnoses for IL-12 receptor Receptor Receptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell. Receptors deficiency: