Fructose metabolism is a complex cascade involving several enzymes Enzymes Enzymes are complex protein biocatalysts that accelerate chemical reactions without being consumed by them. Due to the body's constant metabolic needs, the absence of enzymes would make life unsustainable, as reactions would occur too slowly without these molecules. Basics of Enzymes. The enzymes Enzymes Enzymes are complex protein biocatalysts that accelerate chemical reactions without being consumed by them. Due to the body's constant metabolic needs, the absence of enzymes would make life unsustainable, as reactions would occur too slowly without these molecules. Basics of Enzymes may be deficient or cause abnormal processing and disease. Essential fructosuria, hereditary fructose intolerance, and intestinal fructose intolerance are 3 of the distinct disorders. The presentation may range from asymptomatic to complaints of vomiting Vomiting The forcible expulsion of the contents of the stomach through the mouth. Hypokalemia, bloating Bloating Constipation, flatulence, and diarrhea Diarrhea Diarrhea is defined as ≥ 3 watery or loose stools in a 24-hour period. There are a multitude of etiologies, which can be classified based on the underlying mechanism of disease. The duration of symptoms (acute or chronic) and characteristics of the stools (e.g., watery, bloody, steatorrheic, mucoid) can help guide further diagnostic evaluation. Diarrhea. Management is varied and often focuses on dietary modification. Severe complications such as kidney failure and even death may occur in hereditary fructose intolerance.
Last updated: Apr 25, 2025
Fructose metabolism is an enzymatic cascade, which causes a breakdown of fructose, a monosaccharide, for energy production. The complex process relies upon a series of enzymes Enzymes Enzymes are complex protein biocatalysts that accelerate chemical reactions without being consumed by them. Due to the body’s constant metabolic needs, the absence of enzymes would make life unsustainable, as reactions would occur too slowly without these molecules. Basics of Enzymes (absent in some individuals) and may cause 3 distinct disorders: essential fructosuria, hereditary fructose intolerance, and intestinal fructose intolerance.
Fructolysis is the 1st portion of fructose metabolism:
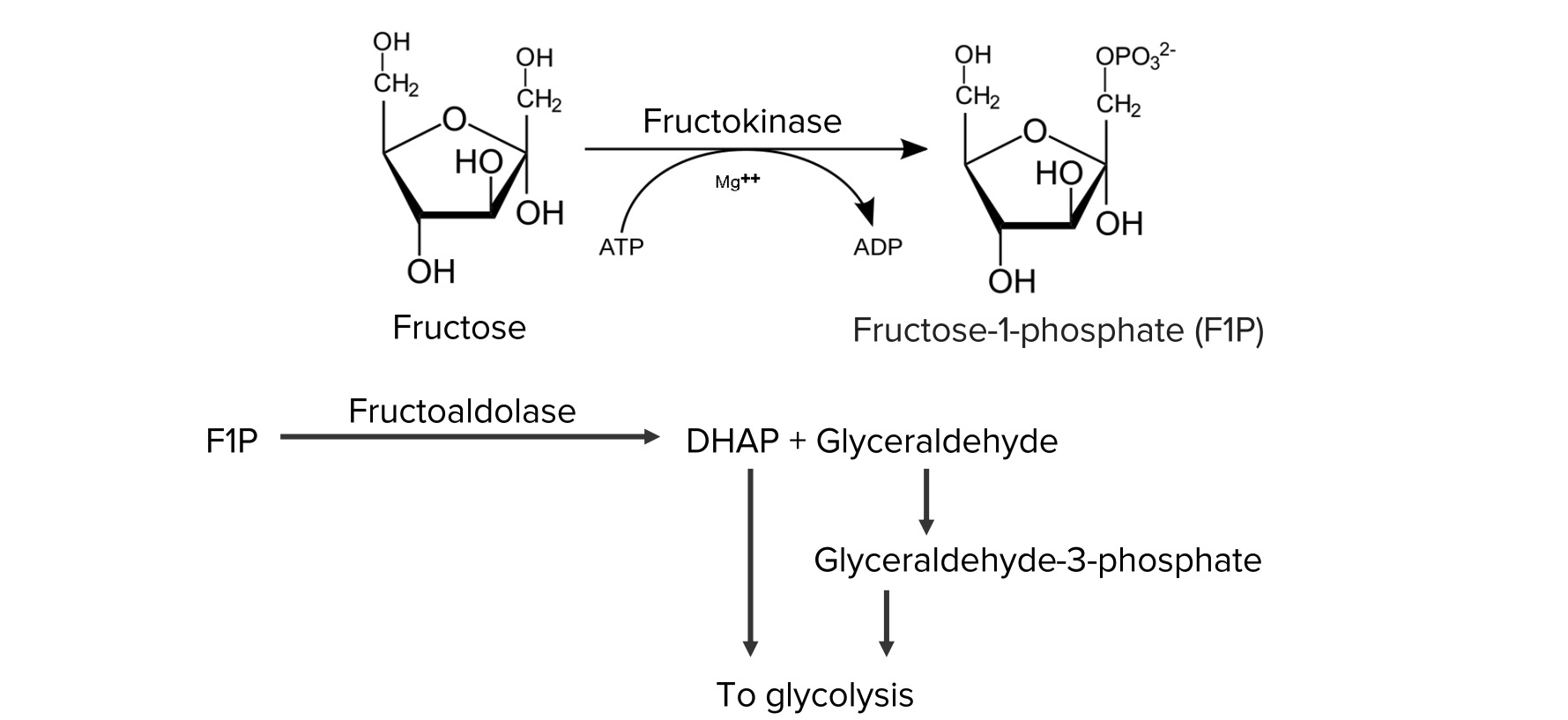
Normal metabolism of fructose:
Fructose is metabolized in hepatocytes to produce dihydroxyacetone phosphate (DHAP) and glyceraldehyde, which are funneled into glycolysis.
Essential fructosuria (fructokinase deficiency):
Hereditary fructose intolerance ( aldolase Aldolase Becker Muscular Dystrophy B/fructoaldolase deficiency):
Intestinal fructose intolerance (deficiency/reduced activity of fructose carriers Carriers The Cell: Cell Membrane of the small intestine Small intestine The small intestine is the longest part of the GI tract, extending from the pyloric orifice of the stomach to the ileocecal junction. The small intestine is the major organ responsible for chemical digestion and absorption of nutrients. It is divided into 3 segments: the duodenum, the jejunum, and the ileum. Small Intestine: Anatomy ( GLUT5 GLUT5 A hexose transporter that mediates fructose transport in skeletal muscle and adipocytes and is responsible for luminal uptake of dietary fructose in the small intestine. Digestion and Absorption)):
Essential fructosuria is asymptomatic.
Symptoms appear when fructose is introduced into the diet.
Symptoms are most evident after ingestion of a fructose load (e.g., juice, fruit).
Hydrogen breath testing detects hydrogen produced by GI bacteria Bacteria Bacteria are prokaryotic single-celled microorganisms that are metabolically active and divide by binary fission. Some of these organisms play a significant role in the pathogenesis of diseases. Bacteriology.
Only patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship with hereditary fructose intolerance have clinically significant complications: