Fatty acid oxidation disorders (FAODs) are a group of genetic conditions caused by disruptions in beta-oxidation or the carnitineCarnitineA constituent of striated muscle and liver. It is an amino acid derivative and an essential cofactor for fatty acid metabolism.Fatty Acid Metabolism transport pathway. These disruptions lead to an inability to metabolize fatty acidsAcidsChemical compounds which yield hydrogen ions or protons when dissolved in water, whose hydrogen can be replaced by metals or basic radicals, or which react with bases to form salts and water (neutralization). An extension of the term includes substances dissolved in media other than water.Acid-Base Balance. All FAOD types are autosomal recessiveAutosomal recessiveAutosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal recessive diseases are only expressed when 2 copies of the recessive allele are inherited.Autosomal Recessive and Autosomal Dominant Inheritance. Because of the inability of the body to break down fatty acidsAcidsChemical compounds which yield hydrogen ions or protons when dissolved in water, whose hydrogen can be replaced by metals or basic radicals, or which react with bases to form salts and water (neutralization). An extension of the term includes substances dissolved in media other than water.Acid-Base Balance, these fatsFatsThe glyceryl esters of a fatty acid, or of a mixture of fatty acids. They are generally odorless, colorless, and tasteless if pure, but they may be flavored according to origin. Fats are insoluble in water, soluble in most organic solvents. They occur in animal and vegetable tissue and are generally obtained by boiling or by extraction under pressure. They are important in the diet (dietary fats) as a source of energy.Energy Homeostasis accumulate in the liverLiverThe liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy and other internal organs. The clinical presentations of each disorder vary, but they commonly include hypoglycemiaHypoglycemiaHypoglycemia is an emergency condition defined as a serum glucose level ≤ 70 mg/dL (≤ 3.9 mmol/L) in diabetic patients. In nondiabetic patients, there is no specific or defined limit for normal serum glucose levels, and hypoglycemia is defined mainly by its clinical features. Hypoglycemia, cardiomyopathyCardiomyopathyCardiomyopathy refers to a group of myocardial diseases associated with structural changes of the heart muscles (myocardium) and impaired systolic and/or diastolic function in the absence of other heart disorders (coronary artery disease, hypertension, valvular disease, and congenital heart disease). Cardiomyopathy: Overview and Types, encephalopathyEncephalopathyHyper-IgM Syndrome, seizuresSeizuresA seizure is abnormal electrical activity of the neurons in the cerebral cortex that can manifest in numerous ways depending on the region of the brain affected. Seizures consist of a sudden imbalance that occurs between the excitatory and inhibitory signals in cortical neurons, creating a net excitation. The 2 major classes of seizures are focal and generalized. Seizures, myopathyMyopathyDermatomyositis, and liverLiverThe liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy dysfunction. ScreeningScreeningPreoperative Care of newborns can detect these diseases, and DNADNAA deoxyribonucleotide polymer that is the primary genetic material of all cells. Eukaryotic and prokaryotic organisms normally contain DNA in a double-stranded state, yet several important biological processes transiently involve single-stranded regions. DNA, which consists of a polysugar-phosphate backbone possessing projections of purines (adenine and guanine) and pyrimidines (thymine and cytosine), forms a double helix that is held together by hydrogen bonds between these purines and pyrimidines (adenine to thymine and guanine to cytosine).DNA Types and Structure sequencing is usually performed to confirm the diagnosis. Management includes dietary changes or substrateSubstrateA substance upon which the enzyme acts.Basics of Enzymes supplementation.
Disorders of fatty acid oxidation are inborn errors of metabolism that disrupt mitochondrial beta-oxidation or fatty acid transportation via the carnitineCarnitineA constituent of striated muscle and liver. It is an amino acid derivative and an essential cofactor for fatty acid metabolism.Fatty Acid Metabolism transport pathway.
Types and classification
Beta-oxidation disorders:
Short-chain acyl-coenzyme A (CoA) dehydrogenase deficiency (SCADD)
Medium-chain acyl-CoA dehydrogenaseAcyl-CoA dehydrogenaseA flavoprotein oxidoreductase that has specificity for medium-chain fatty acids. It forms a complex with electron transferring flavoproteins and conveys reducing equivalents to ubiquinone.Fatty Acid Metabolism deficiency (MCADD)
Very-long-chain acyl-CoA dehydrogenaseAcyl-CoA dehydrogenaseA flavoprotein oxidoreductase that has specificity for medium-chain fatty acids. It forms a complex with electron transferring flavoproteins and conveys reducing equivalents to ubiquinone.Fatty Acid Metabolism deficiency (VLCADD)
Long-chain 3-hydroxy acyl-CoA dehydrogenaseAcyl-CoA dehydrogenaseA flavoprotein oxidoreductase that has specificity for medium-chain fatty acids. It forms a complex with electron transferring flavoproteins and conveys reducing equivalents to ubiquinone.Fatty Acid Metabolism deficiency (LCHADD)
Multiple acyl-CoA dehydrogenaseAcyl-CoA dehydrogenaseA flavoprotein oxidoreductase that has specificity for medium-chain fatty acids. It forms a complex with electron transferring flavoproteins and conveys reducing equivalents to ubiquinone.Fatty Acid Metabolism deficiency (MADD)
3-hydroxy acyl-CoA dehydrogenaseAcyl-CoA dehydrogenaseA flavoprotein oxidoreductase that has specificity for medium-chain fatty acids. It forms a complex with electron transferring flavoproteins and conveys reducing equivalents to ubiquinone.Fatty Acid Metabolism deficiency (HADD)
CarnitineCarnitineA constituent of striated muscle and liver. It is an amino acid derivative and an essential cofactor for fatty acid metabolism.Fatty Acid Metabolism transport disorders:
CarnitineCarnitineA constituent of striated muscle and liver. It is an amino acid derivative and an essential cofactor for fatty acid metabolism.Fatty Acid Metabolism transporter deficiency (CTD)
CarnitineCarnitineA constituent of striated muscle and liver. It is an amino acid derivative and an essential cofactor for fatty acid metabolism.Fatty Acid Metabolism palmitoyltransferase type 1Type 1Spinal Muscular Atrophy deficiency (CPT1D)
CarnitineCarnitineA constituent of striated muscle and liver. It is an amino acid derivative and an essential cofactor for fatty acid metabolism.Fatty Acid Metabolism palmitoyltransferase type 2 deficiency (CPT2D)
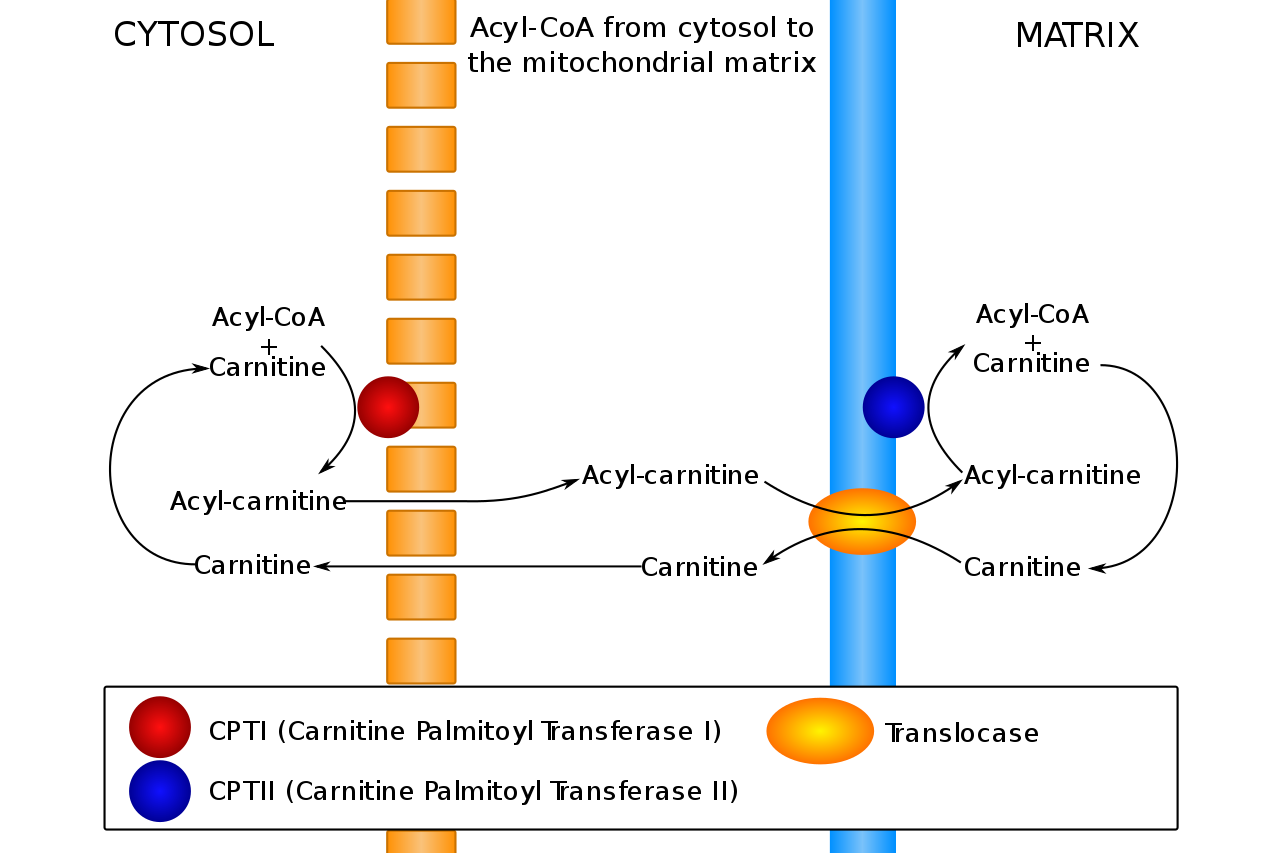
Carnitine transports fatty acids between the cytosol and mitochondrial matrix. Problems with this process lead to disorders of fatty acid metabolism. CoA: coenzyme A
Image: “Acyl-CoA from cytosol to the mitochondrial matrix” by Slagt. License: CC0 1.0
Epidemiology
IncidenceIncidenceThe number of new cases of a given disease during a given period in a specified population. It also is used for the rate at which new events occur in a defined population. It is differentiated from prevalence, which refers to all cases in the population at a given time.Measures of Disease Frequency: approximately 1 in 5000–10,000 live births
The most common is MCADD.
All autosomal recessiveAutosomal recessiveAutosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal recessive diseases are only expressed when 2 copies of the recessive allele are inherited.Autosomal Recessive and Autosomal Dominant Inheritance
Affects both boys and girls
Pathophysiology
The exact pathophysiology varies depending on the deficiency, but there are a couple of overall consequences of disrupting the fatty acid oxidation process:
Reduced energy supply:
Fatty acidsAcidsChemical compounds which yield hydrogen ions or protons when dissolved in water, whose hydrogen can be replaced by metals or basic radicals, or which react with bases to form salts and water (neutralization). An extension of the term includes substances dissolved in media other than water.Acid-Base Balance (FAs) are metabolized for energy in multiple tissues, particularly in a fasting stateFasting stateAbstaining from food.Energy Homeostasis.
An inability to use FAs → body uses glucoseGlucoseA primary source of energy for living organisms. It is naturally occurring and is found in fruits and other parts of plants in its free state. It is used therapeutically in fluid and nutrient replacement.Lactose Intolerance stores normally reserved for vital tissues (e.g., brainBrainThe part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem.Nervous System: Anatomy, Structure, and Classification) → hypoglycemiaHypoglycemiaHypoglycemia is an emergency condition defined as a serum glucose level ≤ 70 mg/dL (≤ 3.9 mmol/L) in diabetic patients. In nondiabetic patients, there is no specific or defined limit for normal serum glucose levels, and hypoglycemia is defined mainly by its clinical features. Hypoglycemia
Some tissues (e.g., skeletal muscle, heart) use FAs in the fed stateFed stateEnergy Homeostasis → loss of this energy source → clinical manifestations
Accumulation of fatty acid metabolites:
Unable to progress through the beta-oxidation process, FAFAInhaled Anesthetics metabolites can build up (particularly within mitochondriaMitochondriaSemiautonomous, self-reproducing organelles that occur in the cytoplasm of all cells of most, but not all, eukaryotes. Each mitochondrion is surrounded by a double limiting membrane. The inner membrane is highly invaginated, and its projections are called cristae. Mitochondria are the sites of the reactions of oxidative phosphorylation, which result in the formation of ATP. They contain distinctive ribosomes, transfer RNAs; amino Acyl tRNA synthetases; and elongation and termination factors. Mitochondria depend upon genes within the nucleus of the cells in which they reside for many essential messenger RNAs. Mitochondria are believed to have arisen from aerobic bacteria that established a symbiotic relationship with primitive protoeukaryotes.The Cell: Organelles)
Some metabolites can be toxic → mitochondrial dysfunction → tissue and organ dysfunction
The presentation and severity varies depending on the disorder. However, there are common signs and symptoms:
Systemic:
FeverFeverFever is defined as a measured body temperature of at least 38°C (100.4°F). Fever is caused by circulating endogenous and/or exogenous pyrogens that increase levels of prostaglandin E2 in the hypothalamus. Fever is commonly associated with chills, rigors, sweating, and flushing of the skin. Fever
Poor appetite
Failure to thriveFailure to ThriveFailure to thrive (FTT), or faltering growth, describes suboptimal weight gain and growth in children. The majority of cases are due to inadequate caloric intake; however, genetic, infectious, and oncological etiologies are also common. Failure to Thrive
Intellectual disabilityDisabilityDetermination of the degree of a physical, mental, or emotional handicap. The diagnosis is applied to legal qualification for benefits and income under disability insurance and to eligibility for social security and workman’s compensation benefits.ABCDE Assessment
SeizuresSeizuresA seizure is abnormal electrical activity of the neurons in the cerebral cortex that can manifest in numerous ways depending on the region of the brain affected. Seizures consist of a sudden imbalance that occurs between the excitatory and inhibitory signals in cortical neurons, creating a net excitation. The 2 major classes of seizures are focal and generalized. Seizures
NauseaNauseaAn unpleasant sensation in the stomach usually accompanied by the urge to vomit. Common causes are early pregnancy, sea and motion sickness, emotional stress, intense pain, food poisoning, and various enteroviruses.Antiemetics and vomitingVomitingThe forcible expulsion of the contents of the stomach through the mouth.Hypokalemia
DiarrheaDiarrheaDiarrhea is defined as ≥ 3 watery or loose stools in a 24-hour period. There are a multitude of etiologies, which can be classified based on the underlying mechanism of disease. The duration of symptoms (acute or chronic) and characteristics of the stools (e.g., watery, bloody, steatorrheic, mucoid) can help guide further diagnostic evaluation. Diarrhea
Hepatic abnormalities are common in infants and children
Hepatomegaly
LiverLiverThe liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy dysfunction
RhabdomyolysisRhabdomyolysisRhabdomyolysis is characterized by muscle necrosis and the release of toxic intracellular contents, especially myoglobin, into the circulation.Rhabdomyolysis may occur in older children and young adults
Cardiac:
CardiomyopathyCardiomyopathyCardiomyopathy refers to a group of myocardial diseases associated with structural changes of the heart muscles (myocardium) and impaired systolic and/or diastolic function in the absence of other heart disorders (coronary artery disease, hypertension, valvular disease, and congenital heart disease). Cardiomyopathy: Overview and Types may be the initial presentation in the neonatal period
Arrhythmia
Other:
HypoglycemiaHypoglycemiaHypoglycemia is an emergency condition defined as a serum glucose level ≤ 70 mg/dL (≤ 3.9 mmol/L) in diabetic patients. In nondiabetic patients, there is no specific or defined limit for normal serum glucose levels, and hypoglycemia is defined mainly by its clinical features. Hypoglycemia
PlasmaPlasmaThe residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation.Transfusion Products acylcarnitine profile
Total and free carnitineCarnitineA constituent of striated muscle and liver. It is an amino acid derivative and an essential cofactor for fatty acid metabolism.Fatty Acid Metabolism levels
DNADNAA deoxyribonucleotide polymer that is the primary genetic material of all cells. Eukaryotic and prokaryotic organisms normally contain DNA in a double-stranded state, yet several important biological processes transiently involve single-stranded regions. DNA, which consists of a polysugar-phosphate backbone possessing projections of purines (adenine and guanine) and pyrimidines (thymine and cytosine), forms a double helix that is held together by hydrogen bonds between these purines and pyrimidines (adenine to thymine and guanine to cytosine).DNA Types and Structure testing provides the final diagnosis
Supportive laboratory evaluation (may vary based on the disorder):
↓ GlucoseGlucoseA primary source of energy for living organisms. It is naturally occurring and is found in fruits and other parts of plants in its free state. It is used therapeutically in fluid and nutrient replacement.Lactose Intolerance and ketonesKetonesOrganic compounds containing a carbonyl group =C=O bonded to two hydrocarbon groups.Basics of Carbohydrates (nonketotic hypoglycemiaHypoglycemiaHypoglycemia is an emergency condition defined as a serum glucose level ≤ 70 mg/dL (≤ 3.9 mmol/L) in diabetic patients. In nondiabetic patients, there is no specific or defined limit for normal serum glucose levels, and hypoglycemia is defined mainly by its clinical features. Hypoglycemia)
Metabolic acidosisAcidosisA pathologic condition of acid accumulation or depletion of base in the body. The two main types are respiratory acidosis and metabolic acidosis, due to metabolic acid build up.Respiratory Acidosis
↑ TransaminasesTransaminasesA subclass of enzymes of the transferase class that catalyze the transfer of an amino group from a donor (generally an amino acid) to an acceptor (generally a 2-keto acid). Most of these enzymes are pyridoxyl phosphate proteins.Autoimmune Hepatitis
↑ AmmoniaAmmoniaA colorless alkaline gas. It is formed in the body during decomposition of organic materials during a large number of metabolically important reactions. Note that the aqueous form of ammonia is referred to as ammonium hydroxide.Acid-Base Balance
↑ CK
Management
Common treatment options include:
Emergency management for decompensation: IV fluidsIV fluidsIntravenous fluids are one of the most common interventions administered in medicine to approximate physiologic bodily fluids. Intravenous fluids are divided into 2 categories: crystalloid and colloid solutions. Intravenous fluids have a wide variety of indications, including intravascular volume expansion, electrolyte manipulation, and maintenance fluids. Intravenous Fluids with dextroseDextroseIntravenous Fluids
Dietary modification:
Avoid fasting.
Snack on low-fat foods.
Eat high-carbohydrate diet.
Supplementation may be helpful in some disorders:
L-carnitineL-carnitineA constituent of striated muscle and liver. It is an amino acid derivative and an essential cofactor for fatty acid metabolism.Drug-Induced Liver Injury
Medium-chain triglyceride oil and triheptanoin are substrateSubstrateA substance upon which the enzyme acts.Basics of Enzymes therapies for long-chain fatty acid oxidation disorders
PrognosisPrognosisA prediction of the probable outcome of a disease based on a individual’s condition and the usual course of the disease as seen in similar situations.Non-Hodgkin Lymphomas
Long-chain fatty acid oxidation disorders are associated with a higher mortalityMortalityAll deaths reported in a given population.Measures of Health Status:
LCHADD
VLCADD
CPT2D
CACTD
Individuals diagnosed through NBS (as opposed to clinically) tend to have better outcomes.
Comparison of Fatty Acid Oxidation Disorders (FAODs)
Table: Comparison of FAODs
Type
Cause
Clinical presentation
CarnitineCarnitineA constituent of striated muscle and liver. It is an amino acid derivative and an essential cofactor for fatty acid metabolism.Fatty Acid Metabolism transport deficiency
(CTD)
MutationMutationGenetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations in SLC22A5
Defective transporter responsible for moving carnitineCarnitineA constituent of striated muscle and liver. It is an amino acid derivative and an essential cofactor for fatty acid metabolism.Fatty Acid Metabolism into cells (deficiency of carnitineCarnitineA constituent of striated muscle and liver. It is an amino acid derivative and an essential cofactor for fatty acid metabolism.Fatty Acid Metabolism in cells)
Difficulty moving FAs into mitochondriaMitochondriaSemiautonomous, self-reproducing organelles that occur in the cytoplasm of all cells of most, but not all, eukaryotes. Each mitochondrion is surrounded by a double limiting membrane. The inner membrane is highly invaginated, and its projections are called cristae. Mitochondria are the sites of the reactions of oxidative phosphorylation, which result in the formation of ATP. They contain distinctive ribosomes, transfer RNAs; amino Acyl tRNA synthetases; and elongation and termination factors. Mitochondria depend upon genes within the nucleus of the cells in which they reside for many essential messenger RNAs. Mitochondria are believed to have arisen from aerobic bacteria that established a symbiotic relationship with primitive protoeukaryotes.The Cell: Organelles
LiverLiverThe liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy dysfunction
HyperammonemiaHyperammonemiaElevated level of ammonia in the blood. It is a sign of defective catabolism of amino acids or ammonia to urea.Cirrhosis
Skeletal muscle weakness
HypoglycemiaHypoglycemiaHypoglycemia is an emergency condition defined as a serum glucose level ≤ 70 mg/dL (≤ 3.9 mmol/L) in diabetic patients. In nondiabetic patients, there is no specific or defined limit for normal serum glucose levels, and hypoglycemia is defined mainly by its clinical features. Hypoglycemia
CardiomyopathyCardiomyopathyCardiomyopathy refers to a group of myocardial diseases associated with structural changes of the heart muscles (myocardium) and impaired systolic and/or diastolic function in the absence of other heart disorders (coronary artery disease, hypertension, valvular disease, and congenital heart disease). Cardiomyopathy: Overview and Types
MutationMutationGenetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations in SLC25A20
Defective carnitine-acylcarnitine translocase
Difficulty moving FAs into mitochondriaMitochondriaSemiautonomous, self-reproducing organelles that occur in the cytoplasm of all cells of most, but not all, eukaryotes. Each mitochondrion is surrounded by a double limiting membrane. The inner membrane is highly invaginated, and its projections are called cristae. Mitochondria are the sites of the reactions of oxidative phosphorylation, which result in the formation of ATP. They contain distinctive ribosomes, transfer RNAs; amino Acyl tRNA synthetases; and elongation and termination factors. Mitochondria depend upon genes within the nucleus of the cells in which they reside for many essential messenger RNAs. Mitochondria are believed to have arisen from aerobic bacteria that established a symbiotic relationship with primitive protoeukaryotes.The Cell: Organelles
SeizuresSeizuresA seizure is abnormal electrical activity of the neurons in the cerebral cortex that can manifest in numerous ways depending on the region of the brain affected. Seizures consist of a sudden imbalance that occurs between the excitatory and inhibitory signals in cortical neurons, creating a net excitation. The 2 major classes of seizures are focal and generalized. Seizures
HypoglycemiaHypoglycemiaHypoglycemia is an emergency condition defined as a serum glucose level ≤ 70 mg/dL (≤ 3.9 mmol/L) in diabetic patients. In nondiabetic patients, there is no specific or defined limit for normal serum glucose levels, and hypoglycemia is defined mainly by its clinical features. Hypoglycemia
Hepatomegaly
HyperammonemiaHyperammonemiaElevated level of ammonia in the blood. It is a sign of defective catabolism of amino acids or ammonia to urea.Cirrhosis
Skeletal muscle weakness
CardiomyopathyCardiomyopathyCardiomyopathy refers to a group of myocardial diseases associated with structural changes of the heart muscles (myocardium) and impaired systolic and/or diastolic function in the absence of other heart disorders (coronary artery disease, hypertension, valvular disease, and congenital heart disease). Cardiomyopathy: Overview and Types
Ventricular arrhythmia
Sudden infant death
CarnitineCarnitineA constituent of striated muscle and liver. It is an amino acid derivative and an essential cofactor for fatty acid metabolism.Fatty Acid Metabolism palmitoyltransferase IA deficiency
(CPT1AD)
MutationMutationGenetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations in CPT1A
Defect in carnitineCarnitineA constituent of striated muscle and liver. It is an amino acid derivative and an essential cofactor for fatty acid metabolism.Fatty Acid Metabolism palmitoyltransferase 1 enzyme
Difficulty moving FAs into mitochondriaMitochondriaSemiautonomous, self-reproducing organelles that occur in the cytoplasm of all cells of most, but not all, eukaryotes. Each mitochondrion is surrounded by a double limiting membrane. The inner membrane is highly invaginated, and its projections are called cristae. Mitochondria are the sites of the reactions of oxidative phosphorylation, which result in the formation of ATP. They contain distinctive ribosomes, transfer RNAs; amino Acyl tRNA synthetases; and elongation and termination factors. Mitochondria depend upon genes within the nucleus of the cells in which they reside for many essential messenger RNAs. Mitochondria are believed to have arisen from aerobic bacteria that established a symbiotic relationship with primitive protoeukaryotes.The Cell: Organelles
HypoglycemiaHypoglycemiaHypoglycemia is an emergency condition defined as a serum glucose level ≤ 70 mg/dL (≤ 3.9 mmol/L) in diabetic patients. In nondiabetic patients, there is no specific or defined limit for normal serum glucose levels, and hypoglycemia is defined mainly by its clinical features. Hypoglycemia
LiverLiverThe liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy dysfunction/failure
Elevated levels of carnitineCarnitineA constituent of striated muscle and liver. It is an amino acid derivative and an essential cofactor for fatty acid metabolism.Fatty Acid Metabolism
CarnitineCarnitineA constituent of striated muscle and liver. It is an amino acid derivative and an essential cofactor for fatty acid metabolism.Fatty Acid Metabolism palmitoyltransferase II (CPT2D)
MutationMutationGenetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations in CPT2
Defect in carnitineCarnitineA constituent of striated muscle and liver. It is an amino acid derivative and an essential cofactor for fatty acid metabolism.Fatty Acid Metabolism palmitoyltransferase 2 enzyme
Difficulty moving FAFAInhaled Anesthetics into mitochondriaMitochondriaSemiautonomous, self-reproducing organelles that occur in the cytoplasm of all cells of most, but not all, eukaryotes. Each mitochondrion is surrounded by a double limiting membrane. The inner membrane is highly invaginated, and its projections are called cristae. Mitochondria are the sites of the reactions of oxidative phosphorylation, which result in the formation of ATP. They contain distinctive ribosomes, transfer RNAs; amino Acyl tRNA synthetases; and elongation and termination factors. Mitochondria depend upon genes within the nucleus of the cells in which they reside for many essential messenger RNAs. Mitochondria are believed to have arisen from aerobic bacteria that established a symbiotic relationship with primitive protoeukaryotes.The Cell: Organelles
Mild to severe adult form:
Exercise intolerance
RhabdomyolysisRhabdomyolysisRhabdomyolysis is characterized by muscle necrosis and the release of toxic intracellular contents, especially myoglobin, into the circulation.Rhabdomyolysis
Renal failureRenal failureConditions in which the kidneys perform below the normal level in the ability to remove wastes, concentrate urine, and maintain electrolyte balance; blood pressure; and calcium metabolism. Renal insufficiency can be classified by the degree of kidney damage (as measured by the level of proteinuria) and reduction in glomerular filtration rate.Crush Syndrome
CardiomyopathyCardiomyopathyCardiomyopathy refers to a group of myocardial diseases associated with structural changes of the heart muscles (myocardium) and impaired systolic and/or diastolic function in the absence of other heart disorders (coronary artery disease, hypertension, valvular disease, and congenital heart disease). Cardiomyopathy: Overview and Types
BrainBrainThe part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem.Nervous System: Anatomy, Structure, and Classification malformations
Death
Multiple acyl-CoA dehydrogenaseAcyl-CoA dehydrogenaseA flavoprotein oxidoreductase that has specificity for medium-chain fatty acids. It forms a complex with electron transferring flavoproteins and conveys reducing equivalents to ubiquinone.Fatty Acid Metabolism deficiency (MADD)
Mutations in ETFA, ETFB, ETFDH
Defect in the electron transport chainElectron transport chainThe electron transport chain (ETC) sends electrons through a series of proteins, which generate an electrochemical proton gradient that produces energy in the form of adenosine triphosphate (ATP).Electron Transport Chain (ETC)
Cannot accept electrons from several acyl-CoA dehydrogenaseAcyl-CoA dehydrogenaseA flavoprotein oxidoreductase that has specificity for medium-chain fatty acids. It forms a complex with electron transferring flavoproteins and conveys reducing equivalents to ubiquinone.Fatty Acid MetabolismenzymesEnzymesEnzymes are complex protein biocatalysts that accelerate chemical reactions without being consumed by them. Due to the body’s constant metabolic needs, the absence of enzymes would make life unsustainable, as reactions would occur too slowly without these molecules. Basics of Enzymes
Disrupts beta-oxidation
Neonatal onset:
Congenital anomalies
Dysmorphic features
CardiomyopathyCardiomyopathyCardiomyopathy refers to a group of myocardial diseases associated with structural changes of the heart muscles (myocardium) and impaired systolic and/or diastolic function in the absence of other heart disorders (coronary artery disease, hypertension, valvular disease, and congenital heart disease). Cardiomyopathy: Overview and Types
Death
Later onset:
VomitingVomitingThe forcible expulsion of the contents of the stomach through the mouth.Hypokalemia
HypoglycemiaHypoglycemiaHypoglycemia is an emergency condition defined as a serum glucose level ≤ 70 mg/dL (≤ 3.9 mmol/L) in diabetic patients. In nondiabetic patients, there is no specific or defined limit for normal serum glucose levels, and hypoglycemia is defined mainly by its clinical features. Hypoglycemia
Metabolic acidosisAcidosisA pathologic condition of acid accumulation or depletion of base in the body. The two main types are respiratory acidosis and metabolic acidosis, due to metabolic acid build up.Respiratory Acidosis
LiverLiverThe liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy dysfunction
Very long-chain acyl-CoA dehydrogenaseAcyl-CoA dehydrogenaseA flavoprotein oxidoreductase that has specificity for medium-chain fatty acids. It forms a complex with electron transferring flavoproteins and conveys reducing equivalents to ubiquinone.Fatty Acid Metabolism deficiency (VLCADD)
MutationMutationGenetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations in ACADVL
Inadequate levels of very-long-chain acyl-CoA dehydrogenaseAcyl-CoA dehydrogenaseA flavoprotein oxidoreductase that has specificity for medium-chain fatty acids. It forms a complex with electron transferring flavoproteins and conveys reducing equivalents to ubiquinone.Fatty Acid Metabolism enzyme
Unable to catalyze initial step in beta-oxidation
HypoglycemiaHypoglycemiaHypoglycemia is an emergency condition defined as a serum glucose level ≤ 70 mg/dL (≤ 3.9 mmol/L) in diabetic patients. In nondiabetic patients, there is no specific or defined limit for normal serum glucose levels, and hypoglycemia is defined mainly by its clinical features. Hypoglycemia
LethargyLethargyA general state of sluggishness, listless, or uninterested, with being tired, and having difficulty concentrating and doing simple tasks. It may be related to depression or drug addiction.Hyponatremia
LiverLiverThe liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy dysfunction
CardiomyopathyCardiomyopathyCardiomyopathy refers to a group of myocardial diseases associated with structural changes of the heart muscles (myocardium) and impaired systolic and/or diastolic function in the absence of other heart disorders (coronary artery disease, hypertension, valvular disease, and congenital heart disease). Cardiomyopathy: Overview and Types
RhabdomyolysisRhabdomyolysisRhabdomyolysis is characterized by muscle necrosis and the release of toxic intracellular contents, especially myoglobin, into the circulation.Rhabdomyolysis
Death
Long-chain 3-hydroxy acyl-CoA dehydrogenaseAcyl-CoA dehydrogenaseA flavoprotein oxidoreductase that has specificity for medium-chain fatty acids. It forms a complex with electron transferring flavoproteins and conveys reducing equivalents to ubiquinone.Fatty Acid Metabolism deficiency
(LCHADD)
Mutations in HADHAgeneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics
Inadequate levels of long-chain 3-hydroxy acyl-CoA dehydrogenaseAcyl-CoA dehydrogenaseA flavoprotein oxidoreductase that has specificity for medium-chain fatty acids. It forms a complex with electron transferring flavoproteins and conveys reducing equivalents to ubiquinone.Fatty Acid Metabolism enzyme
Unable to catalyze step in beta-oxidation
LethargyLethargyA general state of sluggishness, listless, or uninterested, with being tired, and having difficulty concentrating and doing simple tasks. It may be related to depression or drug addiction.Hyponatremia
HypoglycemiaHypoglycemiaHypoglycemia is an emergency condition defined as a serum glucose level ≤ 70 mg/dL (≤ 3.9 mmol/L) in diabetic patients. In nondiabetic patients, there is no specific or defined limit for normal serum glucose levels, and hypoglycemia is defined mainly by its clinical features. Hypoglycemia
LiverLiverThe liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy dysfunction
RhabdomyolysisRhabdomyolysisRhabdomyolysis is characterized by muscle necrosis and the release of toxic intracellular contents, especially myoglobin, into the circulation.Rhabdomyolysis
CardiomyopathyCardiomyopathyCardiomyopathy refers to a group of myocardial diseases associated with structural changes of the heart muscles (myocardium) and impaired systolic and/or diastolic function in the absence of other heart disorders (coronary artery disease, hypertension, valvular disease, and congenital heart disease). Cardiomyopathy: Overview and Types
ComaComaComa is defined as a deep state of unarousable unresponsiveness, characterized by a score of 3 points on the GCS. A comatose state can be caused by a multitude of conditions, making the precise epidemiology and prognosis of coma difficult to determine. Coma
Death
Medium-chain acyl-CoA dehydrogenaseAcyl-CoA dehydrogenaseA flavoprotein oxidoreductase that has specificity for medium-chain fatty acids. It forms a complex with electron transferring flavoproteins and conveys reducing equivalents to ubiquinone.Fatty Acid Metabolism deficiency (MCADD)
MutationMutationGenetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations in ACADM
Inadequate levels of medium-chain acyl-CoA dehydrogenaseAcyl-CoA dehydrogenaseA flavoprotein oxidoreductase that has specificity for medium-chain fatty acids. It forms a complex with electron transferring flavoproteins and conveys reducing equivalents to ubiquinone.Fatty Acid Metabolism enzyme
Unable to catalyze step in beta-oxidation
HypoglycemiaHypoglycemiaHypoglycemia is an emergency condition defined as a serum glucose level ≤ 70 mg/dL (≤ 3.9 mmol/L) in diabetic patients. In nondiabetic patients, there is no specific or defined limit for normal serum glucose levels, and hypoglycemia is defined mainly by its clinical features. Hypoglycemia
LiverLiverThe liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy dysfunction
HyperammonemiaHyperammonemiaElevated level of ammonia in the blood. It is a sign of defective catabolism of amino acids or ammonia to urea.Cirrhosis
LethargyLethargyA general state of sluggishness, listless, or uninterested, with being tired, and having difficulty concentrating and doing simple tasks. It may be related to depression or drug addiction.Hyponatremia
SeizuresSeizuresA seizure is abnormal electrical activity of the neurons in the cerebral cortex that can manifest in numerous ways depending on the region of the brain affected. Seizures consist of a sudden imbalance that occurs between the excitatory and inhibitory signals in cortical neurons, creating a net excitation. The 2 major classes of seizures are focal and generalized. Seizures
Death
Short-chain acyl-CoA dehydrogenaseAcyl-CoA dehydrogenaseA flavoprotein oxidoreductase that has specificity for medium-chain fatty acids. It forms a complex with electron transferring flavoproteins and conveys reducing equivalents to ubiquinone.Fatty Acid Metabolism deficiency (SCADD)
Mutations in ACADS
Deficiency in short-chain acyl-CoA dehydrogenaseAcyl-CoA dehydrogenaseA flavoprotein oxidoreductase that has specificity for medium-chain fatty acids. It forms a complex with electron transferring flavoproteins and conveys reducing equivalents to ubiquinone.Fatty Acid Metabolism enzyme
Unable to catalyze step in beta-oxidation
Asymptomatic
Short-chain 3-hydroxy acyl-CoA dehydrogenaseAcyl-CoA dehydrogenaseA flavoprotein oxidoreductase that has specificity for medium-chain fatty acids. It forms a complex with electron transferring flavoproteins and conveys reducing equivalents to ubiquinone.Fatty Acid Metabolism deficiency (HADD)
MutationMutationGenetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations in HADH
Deficiency in 3-hydroxy acyl-CoA dehydrogenaseAcyl-CoA dehydrogenaseA flavoprotein oxidoreductase that has specificity for medium-chain fatty acids. It forms a complex with electron transferring flavoproteins and conveys reducing equivalents to ubiquinone.Fatty Acid Metabolism enzyme
Unable to catalyze step in beta-oxidation
HypoglycemiaHypoglycemiaHypoglycemia is an emergency condition defined as a serum glucose level ≤ 70 mg/dL (≤ 3.9 mmol/L) in diabetic patients. In nondiabetic patients, there is no specific or defined limit for normal serum glucose levels, and hypoglycemia is defined mainly by its clinical features. Hypoglycemia (hyperinsulinism)
SeizuresSeizuresA seizure is abnormal electrical activity of the neurons in the cerebral cortex that can manifest in numerous ways depending on the region of the brain affected. Seizures consist of a sudden imbalance that occurs between the excitatory and inhibitory signals in cortical neurons, creating a net excitation. The 2 major classes of seizures are focal and generalized. Seizures
Intellectual disabilityDisabilityDetermination of the degree of a physical, mental, or emotional handicap. The diagnosis is applied to legal qualification for benefits and income under disability insurance and to eligibility for social security and workman’s compensation benefits.ABCDE Assessment
FA: fatty acid
References
Actis Dato, V., Milani-Nejad, N., O’Connell, T. D., & Lopaschuk, G. D. (2024). Metabolic flexibility of the heart: The role of fatty acid metabolism in health, heart failure, and cardiometabolic diseases. International Journal of Molecular Sciences, 25(2), 1211. https://doi.org/10.3390/ijms25021211
Blagov, A., Ivanov, A., & Nikiforov, N. (2024). Mitochondrial dysfunction as a factor of energy metabolism disorders in type 2 diabetes mellitus. Frontiers in Bioscience-Scholar, 16(1), 5. https://doi.org/10.31083/j.fbs1601005
Choi, M. G., Lee, J. H., & Park, E. J. (2025). Stabilizing hepatic fatty acid oxidation: Editorial on “USP29 alleviates the progression of MASLD by stabilizing ACSL5 through K48 deubiquitination.” Clinical and Molecular Hepatology, 31(2), 592–595. https://doi.org/10.3350/cmh.2024.0971
Guo, B., Zhang, X., & Wang, Y. (2025). Gut microbiota-derived short chain fatty acids act as mediators of the gut–brain axis targeting age-related neurodegenerative disorders: A narrative review. Critical Reviews in Food Science and Nutrition, 65(2), 265–286. https://doi.org/10.1080/10408398.2023.2272769
Imbard, A., Dionisi-Vici, C., & Houten, S. M. (2025). Circulatory response to exercise relative to oxygen uptake assessed in the follow-up of patients with fatty acid beta-oxidation disorders. Journal of Inherited Metabolic Disease, 48(1), e12819. https://doi.org/10.1002/jimd.12819
Merritt, J. L., MacLeod, E., Jurecka, A., Buhas, D., Dimmock, D., Fürhapter-Rieger, A., & Vockley, J. (2020). Clinical manifestations and management of fatty acid oxidation disorders. Reviews in Endocrine and Metabolic Disorders, 21(4), 479–493. https://doi.org/10.1007/s11154-020-09568-3
Miller, V. R., Bhattacharya, K., & Lee, N. C. (2025). P574: Long-chain fatty acid oxidation disorders: A review of newborn screening around the globe for LC-FAOD. Genetics in Medicine Open, 3, 102417. https://doi.org/10.1016/j.gimo.2025.102417
Mütze, U., Neubauer, B. A., & Wendel, U. (2024). Neurological outcome in long-chain hydroxy fatty acid oxidation disorders. Annals of Clinical and Translational Neurology, 11(4), 883–898. https://doi.org/10.1002/acn3.52002
Sublette, M. E., Ellis, S. P., & Mann, J. J. (2024). The role of polyunsaturated fatty acids in the neurobiology of major depressive disorder and suicide risk. Molecular Psychiatry, 29(2), 269–286. https://doi.org/10.1038/s41380-023-02322-6