Los LOS Neisseria trastornos de oxidación de ácidos grasos son un grupo de afectaciones genéticas causadas por interrupciones en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la beta-oxidación o en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la ruta de transporte de carnitina. Estas interrupciones conducen a una incapacidad para metabolizar los LOS Neisseria ácidos grasos. Todos los LOS Neisseria tipos de trastornos de oxidación de ácidos grasos son autosómicos recesivos. Debido a la incapacidad del cuerpo para descomponer los LOS Neisseria ácidos grasos, estas grasas se acumulan en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el hígado y otros órganos internos. La presentación clínica de cada trastorno varía, pero comúnmente incluyen hipoglucemia, cardiomiopatía, encefalopatía, convulsiones, miopatía y disfunción hepática. La evaluación en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum neonatos puede detectar estas enfermedades y, por lo general, se realiza la secuenciación del ADN para confirmar el diagnóstico. El tratamiento incluye cambios en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la dieta o suplementos de los LOS Neisseria sustratos.
Last updated: Apr 23, 2025
Los LOS Neisseria trastornos de la oxidación de ácidos grasos son errores congénitos del metabolismo que interrumpen la beta-oxidación mitocondrial o el transporte de ácidos grasos a través de la vía de transporte de carnitina.
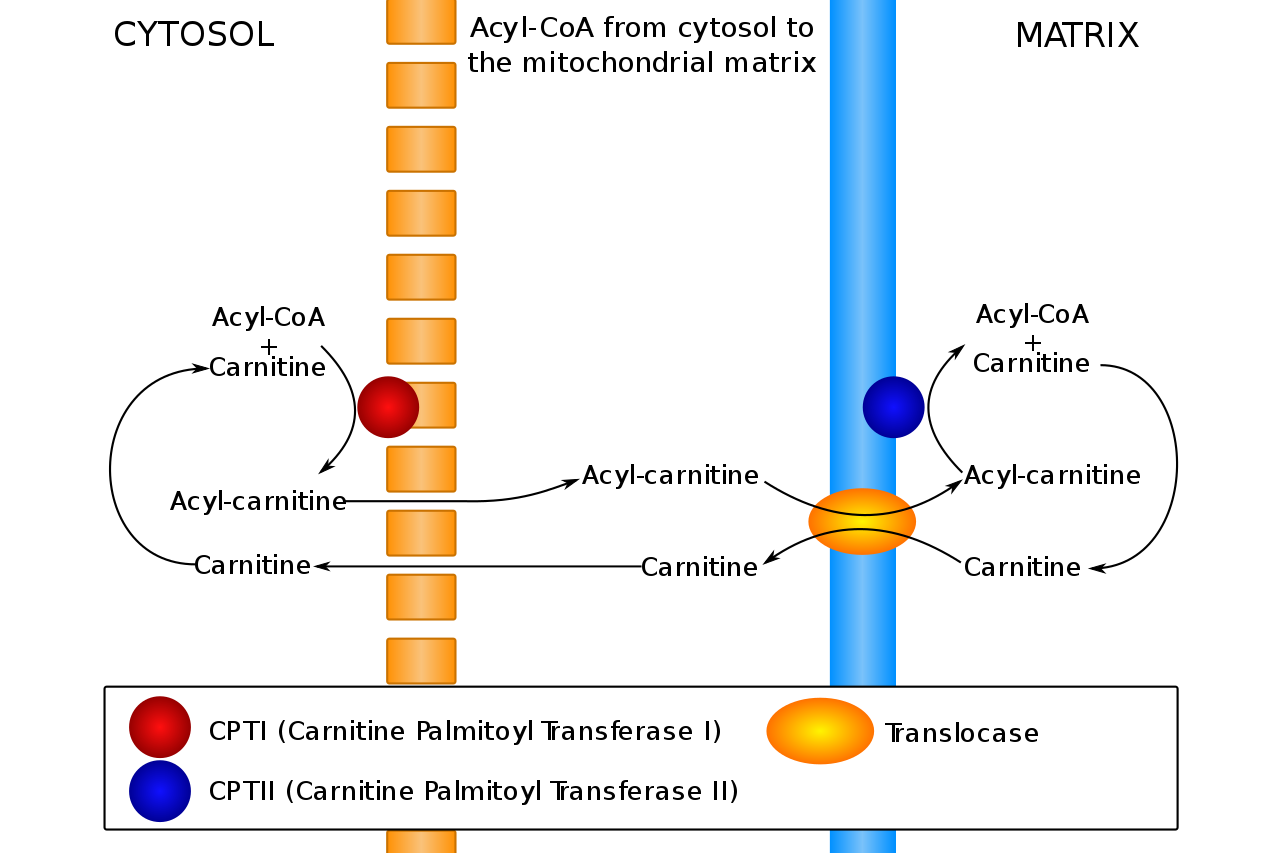
La carnitina transporta ácidos grasos entre el citosol y la matriz mitocondrial. Los problemas con este proceso conducen a trastornos del metabolismo de los ácidos grasos.
CoA: coenzima A
La fisiopatología exacta varía según la deficiencia, pero hay varias consecuencias generales al AL Amyloidosis interrumpir el proceso de oxidación de ácidos grasos:
La presentación y la gravedad varían según el trastorno. Sin embargo, hay signos y síntomas comunes:
Las opciones de tratamiento más frecuente incluye:
| Tipo | Causa | Presentación clínica |
|---|---|---|
| Deficiencia en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el transporte de la carnitina |
|
|
| Deficiencia de carnitina-acilcarnitina translocasa |
|
|
| Deficiencia de carnitina palmitoiltransferasa IA |
|
|
| Carnitina palmitoiltransferasa II |
|
Forma adulta leve a grave:
Forma neonatal:
|
| Deficiencia múltiple de acil-CoA deshidrogenasa |
|
Inicio neonatal:
Inicio tardío:
|
| Deficiencia de acil-CoA deshidrogenasa de cadena muy larga |
|
|
| Deficiencia de 3-hidroxiacil-CoA deshidrogenasa de cadena larga |
|
|
| Deficiencia de acil-CoA deshidrogenasa de cadena media |
|
|
| Deficiencia de acil-CoA deshidrogenasa de cadena corta |
|
Asintomático |
| Deficiencia de 3-hidroxiacil-CoA deshidrogenasa de cadena corta |
|
|