Aromatase Aromatase An enzyme that catalyzes the desaturation (aromatization) of the ring a of C19 androgens and converts them to C18 estrogens. In this process, the 19-methyl is removed. This enzyme is membrane-bound, located in the endoplasmic reticulum of estrogen-producing cells of ovaries, placenta, testes, adipose, and brain tissues. Aromatase is encoded by the cyp19 gene, and functions in complex with NADPH-ferrihemoprotein reductase in the cytochrome p450 system. Adipose Tissue: Histology deficiency is a very rare genetic condition with autosomal recessive inheritance Autosomal recessive inheritance Autosomal Recessive and Autosomal Dominant Inheritance. Aromatase Aromatase An enzyme that catalyzes the desaturation (aromatization) of the ring a of C19 androgens and converts them to C18 estrogens. In this process, the 19-methyl is removed. This enzyme is membrane-bound, located in the endoplasmic reticulum of estrogen-producing cells of ovaries, placenta, testes, adipose, and brain tissues. Aromatase is encoded by the cyp19 gene, and functions in complex with NADPH-ferrihemoprotein reductase in the cytochrome p450 system. Adipose Tissue: Histology deficiency is characterized by congenital estrogen Estrogen Compounds that interact with estrogen receptors in target tissues to bring about the effects similar to those of estradiol. Estrogens stimulate the female reproductive organs, and the development of secondary female sex characteristics. Estrogenic chemicals include natural, synthetic, steroidal, or non-steroidal compounds. Ovaries: Anatomy deprivation with increased levels of testosterone Testosterone A potent androgenic steroid and major product secreted by the leydig cells of the testis. Its production is stimulated by luteinizing hormone from the pituitary gland. In turn, testosterone exerts feedback control of the pituitary LH and FSH secretion. Depending on the tissues, testosterone can be further converted to dihydrotestosterone or estradiol. Androgens and Antiandrogens due to decreased levels of the aromatase Aromatase An enzyme that catalyzes the desaturation (aromatization) of the ring a of C19 androgens and converts them to C18 estrogens. In this process, the 19-methyl is removed. This enzyme is membrane-bound, located in the endoplasmic reticulum of estrogen-producing cells of ovaries, placenta, testes, adipose, and brain tissues. Aromatase is encoded by the cyp19 gene, and functions in complex with NADPH-ferrihemoprotein reductase in the cytochrome p450 system. Adipose Tissue: Histology enzyme. Affected females present with abnormal development of the external genitalia, virilization, primary amenorrhea Amenorrhea Absence of menstruation. Congenital Malformations of the Female Reproductive System, and tall stature. Males usually develop symptoms later in life, including hyperinsulinemia Hyperinsulinemia Diabetes Mellitus and lipid metabolism Lipid Metabolism Lipid metabolism is the processing of lipids for energy use, energy storage, and structural component production. Lipid metabolism uses fats from dietary sources or from fat stores in the body. A complex series of processes involving digestion, absorption, and transport are required for the proper metabolism of lipids. Lipid Metabolism disorders. Individuals affected by aromatase Aromatase An enzyme that catalyzes the desaturation (aromatization) of the ring a of C19 androgens and converts them to C18 estrogens. In this process, the 19-methyl is removed. This enzyme is membrane-bound, located in the endoplasmic reticulum of estrogen-producing cells of ovaries, placenta, testes, adipose, and brain tissues. Aromatase is encoded by the cyp19 gene, and functions in complex with NADPH-ferrihemoprotein reductase in the cytochrome p450 system. Adipose Tissue: Histology deficiency have an increased risk of developing osteoporosis Osteoporosis Osteoporosis refers to a decrease in bone mass and density leading to an increased number of fractures. There are 2 forms of osteoporosis: primary, which is commonly postmenopausal or senile; and secondary, which is a manifestation of immobilization, underlying medical disorders, or long-term use of certain medications. Osteoporosis. Treatment involves hormone replacement therapy Hormone Replacement Therapy Hormone replacement therapy (HRT) is used to treat symptoms associated with female menopause and in combination to suppress ovulation. Risks and side effects include uterine bleeding, predisposition to cancer, breast tenderness, hyperpigmentation, migraine headaches, hypertension, bloating, and mood changes. Noncontraceptive Estrogen and Progestins.
Last updated: Dec 15, 2025
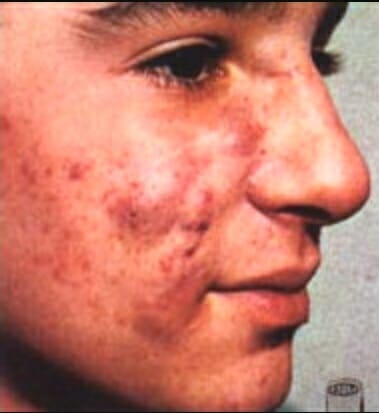
Acne due to virilization during the 12th–30th week of pregnancy
Image: “Retoqueacne” by Jesus Angel Rey. License: Public Domain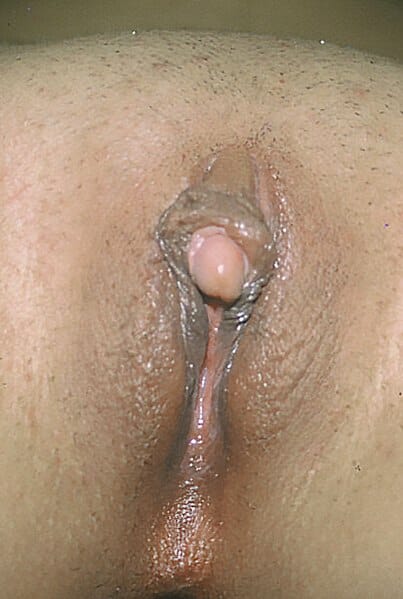
Clitoromegaly, another sign occurring due to virilization
Image: “Clitoromegaly” by Copcu, E, Aktas, A, Sivrioglu, N, Copcu, O, Oztan, Y. License: CC BY 2.0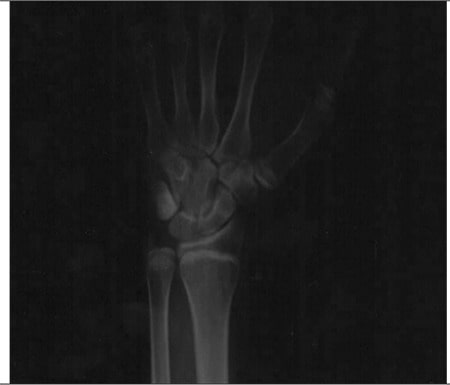
X-ray of a 27-year-old man with aromatase deficiency
Plain bone radiography of the left wrist and hand demonstrates open metacarpal and phalangeal epiphysis. The estimated bone age is 15 years.