


A deficiência de aromatase Aromatase An enzyme that catalyzes the desaturation (aromatization) of the ring a of C19 androgens and converts them to C18 estrogens. In this process, the 19-methyl is removed. This enzyme is membrane-bound, located in the endoplasmic reticulum of estrogen-producing cells of ovaries, placenta, testes, adipose, and brain tissues. Aromatase is encoded by the cyp19 gene, and functions in complex with NADPH-ferrihemoprotein reductase in the cytochrome p450 system. Adipose Tissue: Histology é uma doença genética muito rara com hereditariedade autossómica recessiva. A deficiência de aromatase Aromatase An enzyme that catalyzes the desaturation (aromatization) of the ring a of C19 androgens and converts them to C18 estrogens. In this process, the 19-methyl is removed. This enzyme is membrane-bound, located in the endoplasmic reticulum of estrogen-producing cells of ovaries, placenta, testes, adipose, and brain tissues. Aromatase is encoded by the cyp19 gene, and functions in complex with NADPH-ferrihemoprotein reductase in the cytochrome p450 system. Adipose Tissue: Histology caracteriza-se pela privação congénita de estrogénio com níveis aumentados de testosterona devido à diminuição dos níveis da enzima aromatase Aromatase An enzyme that catalyzes the desaturation (aromatization) of the ring a of C19 androgens and converts them to C18 estrogens. In this process, the 19-methyl is removed. This enzyme is membrane-bound, located in the endoplasmic reticulum of estrogen-producing cells of ovaries, placenta, testes, adipose, and brain tissues. Aromatase is encoded by the cyp19 gene, and functions in complex with NADPH-ferrihemoprotein reductase in the cytochrome p450 system. Adipose Tissue: Histology. As mulheres afetadas apresentam desenvolvimento anormal da genitália externa, virilização, amenorreia primária e estatura alta. Os homens geralmente desenvolvem sintomas numa fase mais MAIS Androgen Insensitivity Syndrome tardia da vida, incluindo hiperinsulinemia e distúrbios do metabolismo lipídico. Os indivíduos afetados pela deficiência de aromatase Aromatase An enzyme that catalyzes the desaturation (aromatization) of the ring a of C19 androgens and converts them to C18 estrogens. In this process, the 19-methyl is removed. This enzyme is membrane-bound, located in the endoplasmic reticulum of estrogen-producing cells of ovaries, placenta, testes, adipose, and brain tissues. Aromatase is encoded by the cyp19 gene, and functions in complex with NADPH-ferrihemoprotein reductase in the cytochrome p450 system. Adipose Tissue: Histology têm um risco aumentado de desenvolver osteoporose. O tratamento envolve terapia de reposição hormonal.
Last updated: Dec 15, 2025
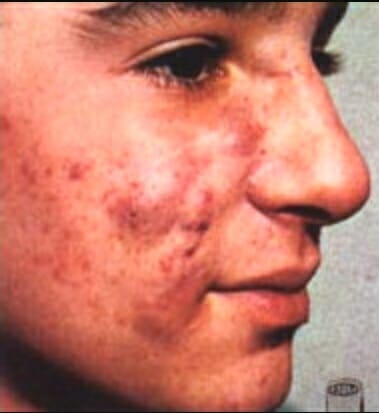
Acne devido à virilização durante a 12ª-30ª semana de gravidez
Imagem: “Retoqueacne” por Jesus Angel Rey. Licença: Public Domain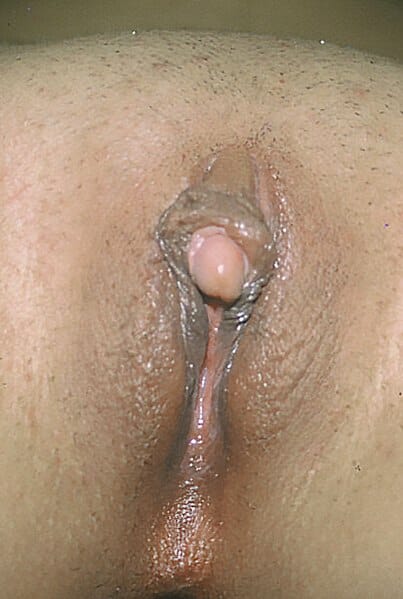
Clitoromegalia, outro sinal que ocorre devido à virilização
Imagem: “Clitoromegaly” por Copcu, E, Aktas, A, Sivrioglu, N, Copcu, O, Oztan, Y. Licença: CC BY 2.0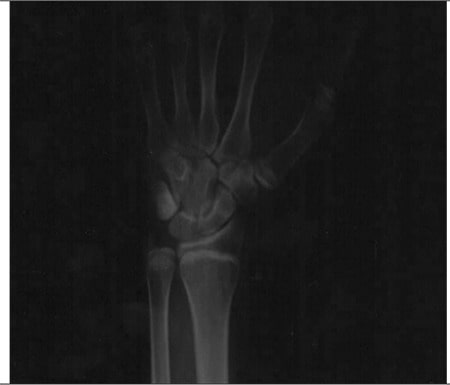
Raio-X de um homem de 27 anos com deficiência de aromatase
A radiografia simples dos ossos do punho e da mão esquerda demonstra epífises metacárpica e falângicas abertas. A idade óssea estimada é de 15 anos.
