Retinoblastoma is a rare tumor Tumor Inflammation but the most common primary intraocular malignancy Malignancy Hemothorax of childhood. It is believed that the condition arises from a neuronal progenitor cell. Retinoblastoma can be heritable or nonheritable. The condition typically presents as unilateral or bilateral leukocoria Leukocoria Cataracts in Children (abnormal white reflection in the eye) in a child under the age of 2. Retinoblastoma is fatal if not treated but early recognition accounts for a high survival rate in resource-rich countries.
Last updated: Dec 15, 2025
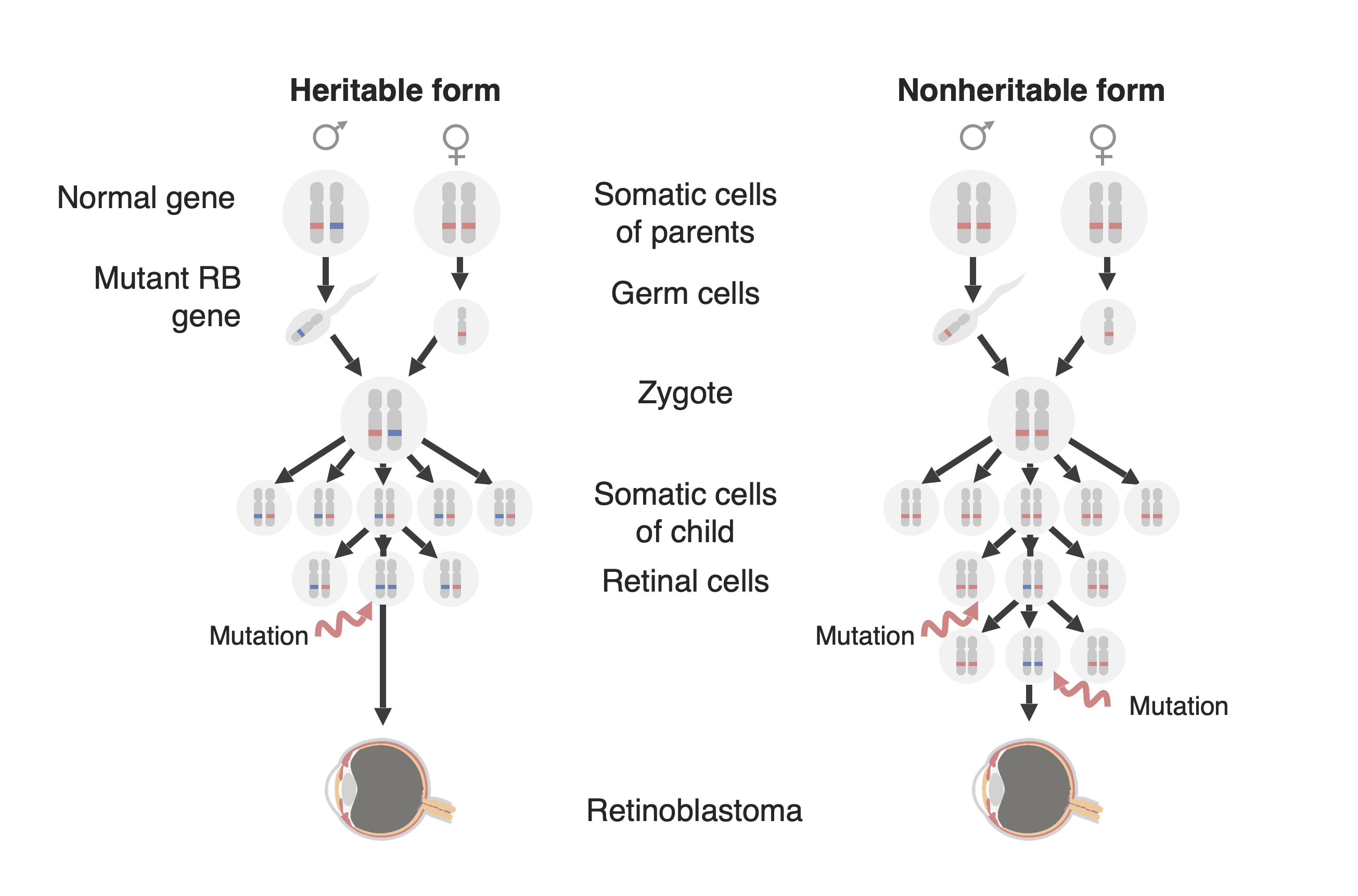
Diagram of the two forms of retinoblastoma development: a familial form (now called the “Heritable form,” on the left and a ”Sporadic form” (now called the “Non-heritable form”) on the right. In the heritable form, a mutated RB1 gene (in blue) is passed on to the child, and only one additional mutation in the paired normal allele (in red) is needed to develop the tumor. In the nonheritable form, the RB1 gene may be mutated de novo in the germline of the offspring (not shown in this figure) so that it then behaves as in the heritable form, or a retinal precursor cell may acquire an RB1 mutation in both alleles and thereby creating a retinoblastoma tumor.
Image by Lecturio.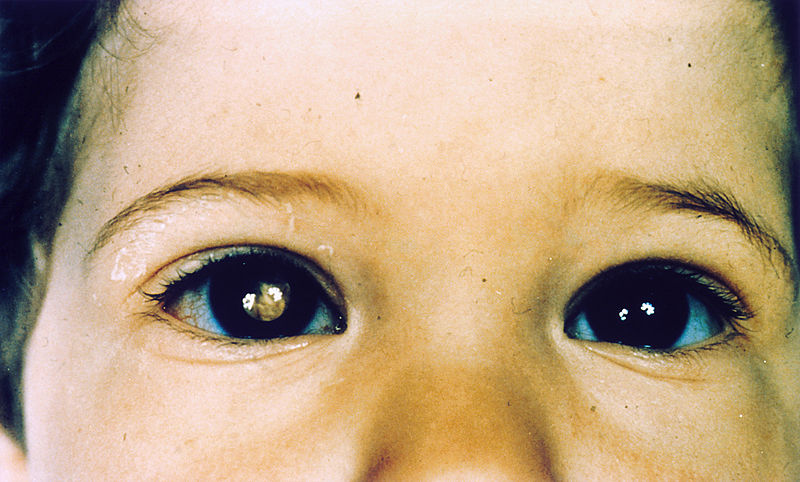
Child with retinoblastoma of the right eye, presenting with leukocoria
Image: “Pathology: Patient: Retinoblastoma” by The National Cancer Institute. License: Public Domain
Child presenting with orbital cellulitis of the right eye caused by locally advanced retinoblastoma
Image: “Orbital cellulitis” by The Pan African Medical Journal. License: CC BY 2.0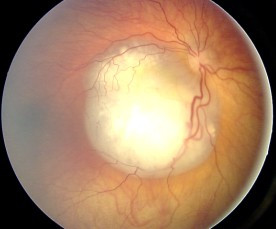
Ocular fundus aspect of retinoblastoma
Image: “Fundus retinoblastoma” by Aerts, I, Lumbroso-Le Rouic. License: CC BY 2.0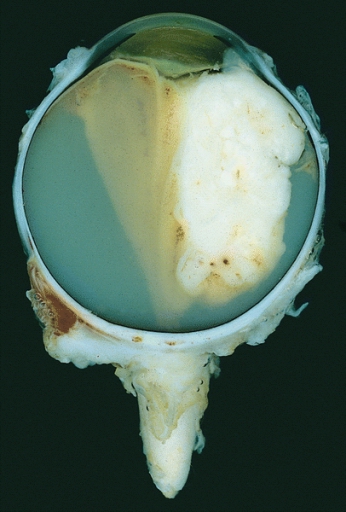
Enucleated eye showing a large exophytic retinoblastoma
Image: “Retinoblastoma” by The Armed Forces Institute of Pathology (AFIP). License: Public Domain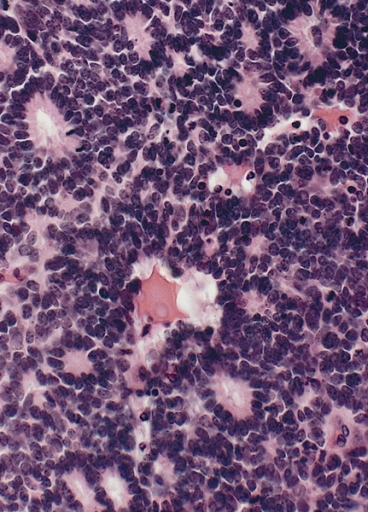
Retinoblastoma with characteristic Flexner-Wintersteiner rosettes featuring a circular alignment of short columnar cells around a central lumen
Image: “Flexner-Wintersteiner rosettes” by The Armed Forces Institute of Pathology (AFIP). License: Public DomainTreatment depends on the stage, with multiple “vision-sparing” therapies available:
The differential diagnosis includes any condition that can cause leukocoria Leukocoria Cataracts in Children.