Pulmonary hypertensionHypertensionHypertension, or high blood pressure, is a common disease that manifests as elevated systemic arterial pressures. Hypertension is most often asymptomatic and is found incidentally as part of a routine physical examination or during triage for an unrelated medical encounter. Hypertension (PHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance) or pulmonary arterial hypertensionHypertensionHypertension, or high blood pressure, is a common disease that manifests as elevated systemic arterial pressures. Hypertension is most often asymptomatic and is found incidentally as part of a routine physical examination or during triage for an unrelated medical encounter. Hypertension (PAHPAHThe glycine amide of 4-aminobenzoic acid. Its sodium salt is used as a diagnostic aid to measure effective renal plasma flow (ERPF) and excretory capacity.Glomerular Filtration) is characterized by elevated pulmonary arterial pressurePulmonary Arterial PressurePulmonary Hypertension Drugs, which can lead to chronic progressive right heart failureHeart FailureA heterogeneous condition in which the heart is unable to pump out sufficient blood to meet the metabolic need of the body. Heart failure can be caused by structural defects, functional abnormalities (ventricular dysfunction), or a sudden overload beyond its capacity. Chronic heart failure is more common than acute heart failure which results from sudden insult to cardiac function, such as myocardial infarction.Total Anomalous Pulmonary Venous Return (TAPVR). Pulmonary hypertensionHypertensionHypertension, or high blood pressure, is a common disease that manifests as elevated systemic arterial pressures. Hypertension is most often asymptomatic and is found incidentally as part of a routine physical examination or during triage for an unrelated medical encounter. Hypertension is grouped into 5 categories based on etiology, which include primary PAHPAHThe glycine amide of 4-aminobenzoic acid. Its sodium salt is used as a diagnostic aid to measure effective renal plasma flow (ERPF) and excretory capacity.Glomerular Filtration, and PHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance due to cardiac disease, lung or hypoxic disease, chronic thromboembolic disease, and multifactorial or unclear etiologies. PatientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship typically present with shortness of breathShortness of breathDyspnea is the subjective sensation of breathing discomfort. Dyspnea is a normal manifestation of heavy physical or psychological exertion, but also may be caused by underlying conditions (both pulmonary and extrapulmonary).Dyspnea initially during exercise and then at rest. Diagnosis may involve an echocardiogramEchocardiogramTransposition of the Great Arteries, ECGECGAn electrocardiogram (ECG) is a graphic representation of the electrical activity of the heart plotted against time. Adhesive electrodes are affixed to the skin surface allowing measurement of cardiac impulses from many angles. The ECG provides 3-dimensional information about the conduction system of the heart, the myocardium, and other cardiac structures. Electrocardiogram (ECG), chest X-rayX-rayPenetrating electromagnetic radiation emitted when the inner orbital electrons of an atom are excited and release radiant energy. X-ray wavelengths range from 1 pm to 10 nm. Hard x-rays are the higher energy, shorter wavelength x-rays. Soft x-rays or grenz rays are less energetic and longer in wavelength. The short wavelength end of the x-ray spectrum overlaps the gamma rays wavelength range. The distinction between gamma rays and x-rays is based on their radiation source.Pulmonary Function Tests, pulmonary function tests, a ventilation-perfusion scan, laboratory testing for conditions associated with PAHPAHThe glycine amide of 4-aminobenzoic acid. Its sodium salt is used as a diagnostic aid to measure effective renal plasma flow (ERPF) and excretory capacity.Glomerular Filtration, and/or cardiac catheterizationCardiac CatheterizationProcedures in which placement of cardiac catheters is performed for therapeutic or diagnostic procedures.Cardiac Surgery. Management is often complex and aimed at treating the underlying etiology. Several classes of vasodilatory agents may be used for patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship with primary PAHPAHThe glycine amide of 4-aminobenzoic acid. Its sodium salt is used as a diagnostic aid to measure effective renal plasma flow (ERPF) and excretory capacity.Glomerular Filtration, including calciumCalciumA basic element found in nearly all tissues. It is a member of the alkaline earth family of metals with the atomic symbol ca, atomic number 20, and atomic weight 40. Calcium is the most abundant mineral in the body and combines with phosphorus to form calcium phosphate in the bones and teeth. It is essential for the normal functioning of nerves and muscles and plays a role in blood coagulation (as factor IV) and in many enzymatic processes.Electrolytes channel blockers and vasoactive prostaglandinsProstaglandinsA group of compounds derived from unsaturated 20-carbon fatty acids, primarily arachidonic acid, via the cyclooxygenase pathway. They are extremely potent mediators of a diverse group of physiological processes.Eicosanoids.
Pulmonary hypertensionHypertensionHypertension, or high blood pressure, is a common disease that manifests as elevated systemic arterial pressures. Hypertension is most often asymptomatic and is found incidentally as part of a routine physical examination or during triage for an unrelated medical encounter. Hypertension (PHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance) is defined as elevated pulmonary arterial pressurePulmonary Arterial PressurePulmonary Hypertension Drugs.
2018 consensus opinion from the 6th World Symposia on Pulmonary HypertensionHypertensionHypertension, or high blood pressure, is a common disease that manifests as elevated systemic arterial pressures. Hypertension is most often asymptomatic and is found incidentally as part of a routine physical examination or during triage for an unrelated medical encounter. Hypertension: mean pulmonary arterial pressurePulmonary Arterial PressurePulmonary Hypertension Drugs > 20 mm Hg at rest
Primary PHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance is referred to as pulmonary arterial hypertensionHypertensionHypertension, or high blood pressure, is a common disease that manifests as elevated systemic arterial pressures. Hypertension is most often asymptomatic and is found incidentally as part of a routine physical examination or during triage for an unrelated medical encounter. Hypertension (PAHPAHThe glycine amide of 4-aminobenzoic acid. Its sodium salt is used as a diagnostic aid to measure effective renal plasma flow (ERPF) and excretory capacity.Glomerular Filtration).
Secondary PHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance may be due to chronic heart, lung, or systemic diseases.
Epidemiology
Secondary PHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance is much more common than the primary variant. The epidemiology of secondary PHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance is similar to the underlying condition.
Primary PHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance/PAHPAHThe glycine amide of 4-aminobenzoic acid. Its sodium salt is used as a diagnostic aid to measure effective renal plasma flow (ERPF) and excretory capacity.Glomerular Filtration:
PrevalencePrevalenceThe total number of cases of a given disease in a specified population at a designated time. It is differentiated from incidence, which refers to the number of new cases in the population at a given time.Measures of Disease Frequency: approximately 15 cases in 1 million adults
SchistosomiasisSchistosomiasisInfection with flukes (trematodes) of the genus schistosoma. Three species produce the most frequent clinical diseases: Schistosoma haematobium (endemic in Africa and the Middle East), Schistosoma Mansoni (in Egypt, northern and southern Africa, some West Indies islands, northern 2/3 of South america), and Schistosoma japonicum (in Japan, China, the Philippines, Celebes, Thailand, Laos). S. mansoni is often seen in Puerto Ricans living in the United States.Schistosoma/Schistosomiasis is the most common cause worldwide.
In areas without endemic schistosomiasisSchistosomiasisInfection with flukes (trematodes) of the genus schistosoma. Three species produce the most frequent clinical diseases: Schistosoma haematobium (endemic in Africa and the Middle East), Schistosoma Mansoni (in Egypt, northern and southern Africa, some West Indies islands, northern 2/3 of South america), and Schistosoma japonicum (in Japan, China, the Philippines, Celebes, Thailand, Laos). S. mansoni is often seen in Puerto Ricans living in the United States.Schistosoma/Schistosomiasis:
GenderGenderGender DysphoriabiasBiasEpidemiological studies are designed to evaluate a hypothesized relationship between an exposure and an outcome; however, the existence and/or magnitude of these relationships may be erroneously affected by the design and execution of the study itself or by conscious or unconscious errors perpetrated by the investigators or the subjects. These systematic errors are called biases. Types of Biases: women > men
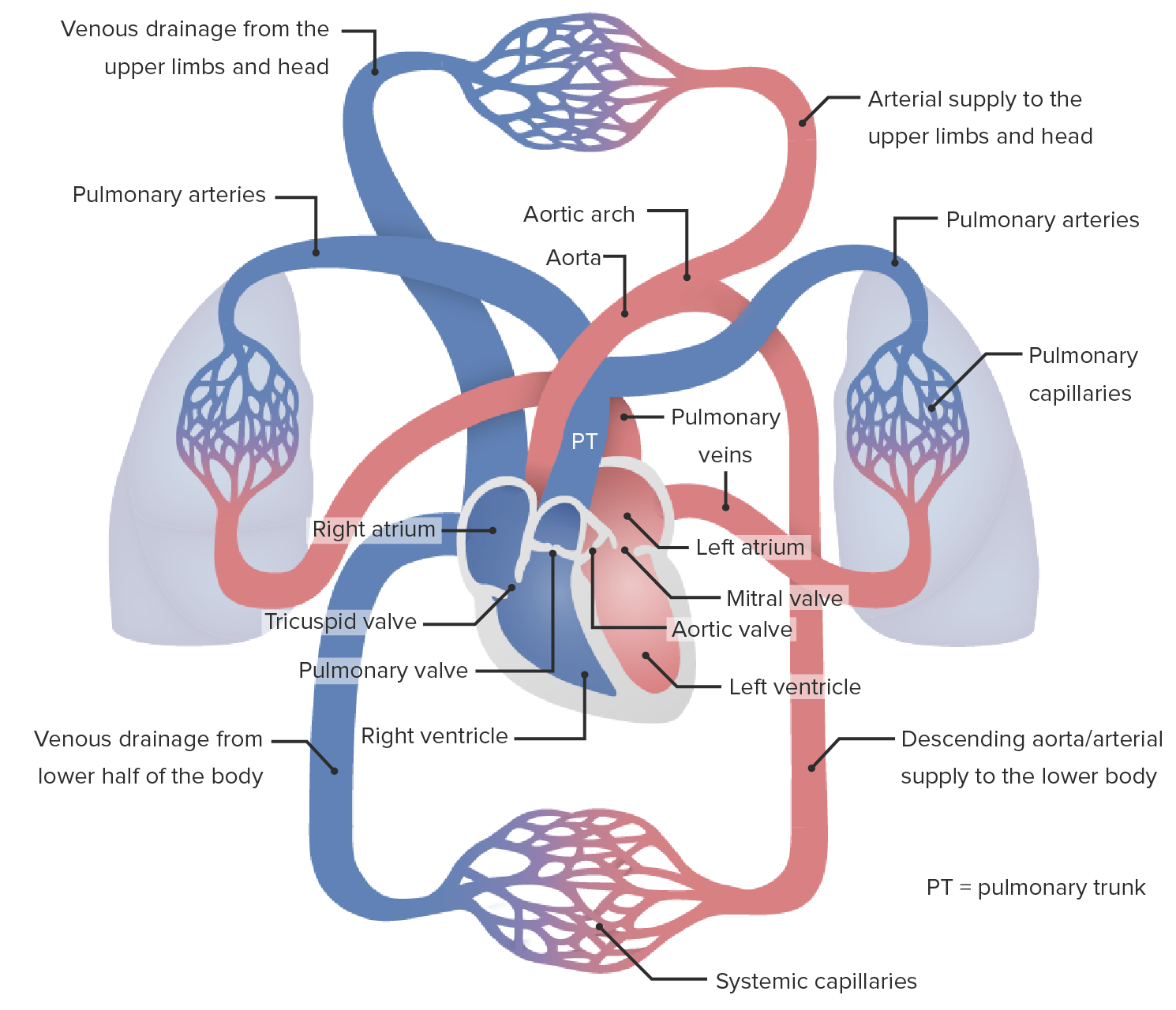
Blood flowBlood flowBlood flow refers to the movement of a certain volume of blood through the vasculature over a given unit of time (e.g., mL per minute).Vascular Resistance, Flow, and Mean Arterial Pressure through the cardiac and pulmonary circulationCirculationThe movement of the blood as it is pumped through the cardiovascular system.ABCDE Assessment takes the following path:
Deoxygenated systemic blood returns to the right side of the heart through the superior and inferior venae cavaeVenae CavaeThe inferior and superior venae cavae.Veins: Histology.
Vena cava → right atrium → tricuspid valveTricuspid valveThe valve consisting of three cusps situated between the right atrium and right ventricle of the heart.Heart: Anatomy → right ventricle → pulmonary valvePulmonary valveA valve situated at the entrance to the pulmonary trunk from the right ventricle.Heart: Anatomy → pulmonary trunkPulmonary TrunkTruncus Arteriosus
Pulmonary arteriesArteriesArteries are tubular collections of cells that transport oxygenated blood and nutrients from the heart to the tissues of the body. The blood passes through the arteries in order of decreasing luminal diameter, starting in the largest artery (the aorta) and ending in the small arterioles. Arteries are classified into 3 types: large elastic arteries, medium muscular arteries, and small arteries and arterioles. Arteries: Histology → pulmonary capillariesCapillariesCapillaries are the primary structures in the circulatory system that allow the exchange of gas, nutrients, and other materials between the blood and the extracellular fluid (ECF). Capillaries are the smallest of the blood vessels. Because a capillary diameter is so small, only 1 RBC may pass through at a time.Capillaries: Histology (blood is oxygenated) → pulmonary veinsPulmonary veinsThe veins that return the oxygenated blood from the lungs to the left atrium of the heart.Lungs: Anatomy
Oxygenated blood returns to the left side of the heart.
Pulmonary veinsPulmonary veinsThe veins that return the oxygenated blood from the lungs to the left atrium of the heart.Lungs: Anatomy → left atrium → mitral valveMitral valveThe valve between the left atrium and left ventricle of the heart.Heart: Anatomy → left ventricle → aortic valveAortic valveThe valve between the left ventricle and the ascending aorta which prevents backflow into the left ventricle.Heart: Anatomy → aortaAortaThe main trunk of the systemic arteries.Mediastinum and Great Vessels: Anatomy
The WHO has classified PHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance into 5 categories based on the etiology:
Group 1: PAHPAHThe glycine amide of 4-aminobenzoic acid. Its sodium salt is used as a diagnostic aid to measure effective renal plasma flow (ERPF) and excretory capacity.Glomerular Filtration
Group 1 refers to cases of increased pulmonary arterial pressurePulmonary Arterial PressurePulmonary Hypertension Drugsin the absence of underlying heart or lung disease and was previously called (and can still be conceptualized as) primaryPHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance. The etiologies of group 1 include:
IdiopathicIdiopathicDermatomyositisPAHPAHThe glycine amide of 4-aminobenzoic acid. Its sodium salt is used as a diagnostic aid to measure effective renal plasma flow (ERPF) and excretory capacity.Glomerular Filtration (most common cause of PAHPAHThe glycine amide of 4-aminobenzoic acid. Its sodium salt is used as a diagnostic aid to measure effective renal plasma flow (ERPF) and excretory capacity.Glomerular Filtration)
Familial PAHPAHThe glycine amide of 4-aminobenzoic acid. Its sodium salt is used as a diagnostic aid to measure effective renal plasma flow (ERPF) and excretory capacity.Glomerular Filtration: due to mutations in the boneBoneBone is a compact type of hardened connective tissue composed of bone cells, membranes, an extracellular mineralized matrix, and central bone marrow. The 2 primary types of bone are compact and spongy. Bones: Structure and Types morphogenic protein-receptor-2 (BMPR2) geneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics, several other rare mutations, or unknown causes
Drugs or toxins:
Appetite suppressants:
AmphetaminesAmphetaminesAnalogs or derivatives of amphetamine. Many are sympathomimetics and central nervous system stimulators causing excitation, vasopressin, bronchodilation, and to varying degrees, anorexia, analepsis, nasal decongestion, and some smooth muscle relaxation.Stimulants
Fenfluramine
Aminorex
Rapeseed oil
CocaineCocaineAn alkaloid ester extracted from the leaves of plants including coca. It is a local anesthetic and vasoconstrictor and is clinically used for that purpose, particularly in the eye, ear, nose, and throat. It also has powerful central nervous system effects similar to the amphetamines and is a drug of abuse. Cocaine, like amphetamines, acts by multiple mechanisms on brain catecholaminergic neurons; the mechanism of its reinforcing effects is thought to involve inhibition of dopamine uptake.Local Anesthetics
Associated with:
Connective tissueConnective tissueConnective tissues originate from embryonic mesenchyme and are present throughout the body except inside the brain and spinal cord. The main function of connective tissues is to provide structural support to organs. Connective tissues consist of cells and an extracellular matrix.Connective Tissue: Histology disease (primarily sclerodermaSclerodermaScleroderma (systemic sclerosis) is an autoimmune condition characterized by diffuse collagen deposition and fibrosis. The clinical presentation varies from limited skin involvement to diffuse involvement of internal organs. Scleroderma)
Portal hypertensionPortal hypertensionPortal hypertension is increased pressure in the portal venous system. This increased pressure can lead to splanchnic vasodilation, collateral blood flow through portosystemic anastomoses, and increased hydrostatic pressure. There are a number of etiologies, including cirrhosis, right-sided congestive heart failure, schistosomiasis, portal vein thrombosis, hepatitis, and Budd-Chiari syndrome. Portal Hypertension
SchistosomiasisSchistosomiasisInfection with flukes (trematodes) of the genus schistosoma. Three species produce the most frequent clinical diseases: Schistosoma haematobium (endemic in Africa and the Middle East), Schistosoma Mansoni (in Egypt, northern and southern Africa, some West Indies islands, northern 2/3 of South america), and Schistosoma japonicum (in Japan, China, the Philippines, Celebes, Thailand, Laos). S. mansoni is often seen in Puerto Ricans living in the United States.Schistosoma/Schistosomiasis (among the most common causes of PAHPAHThe glycine amide of 4-aminobenzoic acid. Its sodium salt is used as a diagnostic aid to measure effective renal plasma flow (ERPF) and excretory capacity.Glomerular Filtration worldwide)
Group 2: PHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance due to left heart disease
Left-sided heart disease can cause pressure backup through the pulmonary vasculature leading to PHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance, and is the most common cause of PHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance overall. Etiologies include:
Left-sided atrial or ventricular heart disease
Left-sided valvular heart disease
Obstruction of the left heart inflow or outflow tracts (congenitalCongenitalChorioretinitis or acquired)
CongenitalCongenitalChorioretinitiscardiomyopathiesCardiomyopathiesA group of diseases in which the dominant feature is the involvement of the cardiac muscle itself. Cardiomyopathies are classified according to their predominant pathophysiological features (dilated cardiomyopathy; hypertrophic cardiomyopathy; restrictive cardiomyopathy) or their etiological/pathological factors (cardiomyopathy, alcoholic; endocardial fibroelastosis).Cardiomyopathy: Overview and Types
Group 3: PHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance due to chronic lung disease and/or hypoxiaHypoxiaSub-optimal oxygen levels in the ambient air of living organisms.Ischemic Cell Damage
HypoxiaHypoxiaSub-optimal oxygen levels in the ambient air of living organisms.Ischemic Cell Damage leads to physiological vasoconstrictionVasoconstrictionThe physiological narrowing of blood vessels by contraction of the vascular smooth muscle.Vascular Resistance, Flow, and Mean Arterial Pressure of the pulmonary vasculature to prevent ventilation-perfusion mismatch. As a result, chronic hypoxiaHypoxiaSub-optimal oxygen levels in the ambient air of living organisms.Ischemic Cell Damage, as well as destructive lung diseases, can lead to chronic PHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance. Etiologies include:
Chronic obstructive pulmonary diseasePulmonary diseaseDiseases involving the respiratory system.Blastomyces/Blastomycosis (COPDCOPDChronic obstructive pulmonary disease (COPD) is a lung disease characterized by progressive, largely irreversible airflow obstruction. The condition usually presents in middle-aged or elderly persons with a history of cigarette smoking. Signs and symptoms include prolonged expiration, wheezing, diminished breath sounds, progressive dyspnea, and chronic cough. Chronic Obstructive Pulmonary Disease (COPD)) (most common)
Interstitial lung disease
Pulmonary diseases with mixed restrictive and obstructive patterns
Obesity hypoventilation syndromeObesity hypoventilation syndromeHypoventilation syndrome in very obese persons with excessive adipose tissue around the abdomen and diaphragm. It is characterized by diminished to absent ventilatory chemoresponsiveness; chronic hypoxia; hypercapnia; polycythemia; and long periods of sleep during day and night (hypersomnolence). It is a condition often related to obstructive sleep apnea but can occur separately.Obstructive Sleep Apnea
Group 4: PHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance due to chronic thromboembolic disease
Group 4 cases are diagnosed when there is an increase in pulmonary arterial pressurePulmonary Arterial PressurePulmonary Hypertension Drugs with documentationDocumentationSystematic organization, storage, retrieval, and dissemination of specialized information, especially of a scientific or technical nature. It often involves authenticating or validating information.Advance Directives of pulmonary arterial obstruction. Etiologies include:
Nonthrombotic pulmonary embolismPulmonary EmbolismPulmonary embolism (PE) is a potentially fatal condition that occurs as a result of intraluminal obstruction of the main pulmonary artery or its branches. The causative factors include thrombi, air, amniotic fluid, and fat. In PE, gas exchange is impaired due to the decreased return of deoxygenated blood to the lungs. Pulmonary Embolism:
TumorTumorInflammation embolisms (late-stage manifestations of certain malignancies with embolizationEmbolizationA method of hemostasis utilizing various agents such as gelfoam, silastic, metal, glass, or plastic pellets, autologous clot, fat, and muscle as emboli. It has been used in the treatment of spinal cord and intracranial arteriovenous malformations, renal arteriovenous fistulas, gastrointestinal bleeding, epistaxis, hypersplenism, certain highly vascular tumors, traumatic rupture of blood vessels, and control of operative hemorrhage.Gastrointestinal Bleeding of tumorTumorInflammation particles themselves)
Foreign material (most commonly silicone, after surgical injection)
Group 5: PHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance due to unclear or multifactorial causes
Pulmonary hypertensionHypertensionHypertension, or high blood pressure, is a common disease that manifests as elevated systemic arterial pressures. Hypertension is most often asymptomatic and is found incidentally as part of a routine physical examination or during triage for an unrelated medical encounter. Hypertension is classified as Group 5 when the elevation in pulmonary arterial pressurePulmonary Arterial PressurePulmonary Hypertension Drugs is associated with a systemic disease, where a causal relationship is not clearly understood or thought to be multifactorial. Etiologies include:
Hematological disorders:
Chronic hemolytic anemias:
Sickle cell diseaseSickle cell diseaseSickle cell disease (SCD) is a group of genetic disorders in which an abnormal Hb molecule (HbS) transforms RBCs into sickle-shaped cells, resulting in chronic anemia, vasoocclusive episodes, pain, and organ damage.Sickle Cell Disease
ThalassemiaThalassemiaThalassemia is a hereditary cause of microcytic hypochromic anemia and results from a deficiency in either the α or β globin chains, resulting in hemoglobinopathy. The presentation of thalassemia depends on the number of defective chains present and can range from being asymptomatic to rendering the more severely affected patients to be transfusion dependent. Thalassemia
SarcoidosisSarcoidosisSarcoidosis is a multisystem inflammatory disease that causes noncaseating granulomas. The exact etiology is unknown. Sarcoidosis usually affects the lungs and thoracic lymph nodes, but it can also affect almost every system in the body, including the skin, heart, and eyes, most commonly. Sarcoidosis
Pulmonary histiocytosis
LymphangioleiomyomatosisLymphangioleiomyomatosisA disease characterized by the progressive invasion of smooth muscle cells into the lymphatic vessels, and the blood vessels. The majority of the cases occur in the lungs of women of childbearing age, eventually blocking the flow of air, blood, and lymph. The common symptom is shortness of breath (dyspnea).Interstitial Lung Diseases
Metabolic disorders:
Glycogen storage diseaseGlycogen storage diseaseA group of inherited metabolic disorders involving the enzymes responsible for the synthesis and degradation of glycogen. In some patients, prominent liver involvement is presented. In others, more generalized storage of glycogen occurs, sometimes with prominent cardiac involvement.Benign Liver Tumors
Gaucher diseaseGaucher diseaseGaucher Disease (GD) is an autosomal recessive lysosomal storage disorder caused by a deficiency of glucocerebrosidase enzyme activity, resulting in accumulation of glucocerebroside in cells and certain organs. The disease is categorized into 3 types with variable clinical presentation. Gaucher Disease
ThyroidThyroidThe thyroid gland is one of the largest endocrine glands in the human body. The thyroid gland is a highly vascular, brownish-red gland located in the visceral compartment of the anterior region of the neck.Thyroid Gland: Anatomy disorders
Chronic renal failureRenal failureConditions in which the kidneys perform below the normal level in the ability to remove wastes, concentrate urine, and maintain electrolyte balance; blood pressure; and calcium metabolism. Renal insufficiency can be classified by the degree of kidney damage (as measured by the level of proteinuria) and reduction in glomerular filtration rate.Crush Syndrome
Enlarged proximal pulmonary arteriesArteriesArteries are tubular collections of cells that transport oxygenated blood and nutrients from the heart to the tissues of the body. The blood passes through the arteries in order of decreasing luminal diameter, starting in the largest artery (the aorta) and ending in the small arterioles. Arteries are classified into 3 types: large elastic arteries, medium muscular arteries, and small arteries and arterioles. Arteries: Histology
Ultimately results in cor pulmonaleCor PulmonaleCor pulmonale is right ventricular (RV) dysfunction caused by lung disease that results in pulmonary artery hypertension. The most common cause of cor pulmonale is chronic obstructive pulmonary disease. Dyspnea is the usual presenting symptom. Cor Pulmonale(right-sided heart failureRight-Sided Heart FailureEbstein’s Anomaly).
Q: right-sided cardiac outputCardiac outputThe volume of blood passing through the heart per unit of time. It is usually expressed as liters (volume) per minute so as not to be confused with stroke volume (volume per beat).Cardiac Mechanics
PVR: pulmonary vascular resistanceResistancePhysiologically, the opposition to flow of air caused by the forces of friction. As a part of pulmonary function testing, it is the ratio of driving pressure to the rate of air flow.Ventilation: Mechanics of Breathing
Pulmonary capillary wedge pressure estimates left atrial pressure.
↑ Pulmonary vascular resistanceResistancePhysiologically, the opposition to flow of air caused by the forces of friction. As a part of pulmonary function testing, it is the ratio of driving pressure to the rate of air flow.Ventilation: Mechanics of Breathing (most common)
↑ FlowFlowBlood flows through the heart, arteries, capillaries, and veins in a closed, continuous circuit. Flow is the movement of volume per unit of time. Flow is affected by the pressure gradient and the resistance fluid encounters between 2 points. Vascular resistance is the opposition to flow, which is caused primarily by blood friction against vessel walls.Vascular Resistance, Flow, and Mean Arterial Pressure through the pulmonary vasculature
↑ Left-sided pressures
Increased pulmonary vascular resistanceResistancePhysiologically, the opposition to flow of air caused by the forces of friction. As a part of pulmonary function testing, it is the ratio of driving pressure to the rate of air flow.Ventilation: Mechanics of Breathing
Increased pulmonary vascular resistanceResistancePhysiologically, the opposition to flow of air caused by the forces of friction. As a part of pulmonary function testing, it is the ratio of driving pressure to the rate of air flow.Ventilation: Mechanics of Breathingis the primary cause of PHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance in most cases and may be due to:
Occlusive vasculopathies of the small pulmonary arteriesArteriesArteries are tubular collections of cells that transport oxygenated blood and nutrients from the heart to the tissues of the body. The blood passes through the arteries in order of decreasing luminal diameter, starting in the largest artery (the aorta) and ending in the small arterioles. Arteries are classified into 3 types: large elastic arteries, medium muscular arteries, and small arteries and arterioles. Arteries: Histology/arteriolesArteriolesThe smallest divisions of the arteries located between the muscular arteries and the capillaries.Arteries: Histology: remodel the vasculature and alter the tone (e.g., idiopathicIdiopathicDermatomyositisPAHPAHThe glycine amide of 4-aminobenzoic acid. Its sodium salt is used as a diagnostic aid to measure effective renal plasma flow (ERPF) and excretory capacity.Glomerular Filtration)
Mitral valveMitral valveThe valve between the left atrium and left ventricle of the heart.Heart: Anatomy disease
Left ventricular dysfunction
Constrictive pericarditisConstrictive pericarditisInflammation of the pericardium that is characterized by the fibrous scarring and adhesion of both serous layers, the visceral pericardium and the parietal pericardium leading to the loss of pericardial cavity. The thickened pericardium severely restricts cardiac filling. Clinical signs include fatigue, muscle wasting, and weight loss.Pericarditis
Restrictive cardiomyopathyRestrictive CardiomyopathyRestrictive cardiomyopathy (RCM) is a fairly uncommon condition characterized by progressive stiffening of the cardiac muscle, which causes impaired relaxation and refilling of the heart during diastole, resulting in diastolic dysfunction and eventual heart failure. Restrictive Cardiomyopathy
Pulmonary venous obstruction
Increased flowFlowBlood flows through the heart, arteries, capillaries, and veins in a closed, continuous circuit. Flow is the movement of volume per unit of time. Flow is affected by the pressure gradient and the resistance fluid encounters between 2 points. Vascular resistance is the opposition to flow, which is caused primarily by blood friction against vessel walls.Vascular Resistance, Flow, and Mean Arterial Pressure through the pulmonary vasculature
Typically, increased flowFlowBlood flows through the heart, arteries, capillaries, and veins in a closed, continuous circuit. Flow is the movement of volume per unit of time. Flow is affected by the pressure gradient and the resistance fluid encounters between 2 points. Vascular resistance is the opposition to flow, which is caused primarily by blood friction against vessel walls.Vascular Resistance, Flow, and Mean Arterial Pressure triggers vasodilationVasodilationThe physiological widening of blood vessels by relaxing the underlying vascular smooth muscle.Pulmonary Hypertension Drugs of the pulmonary vasculature. In cases where this increase in flowFlowBlood flows through the heart, arteries, capillaries, and veins in a closed, continuous circuit. Flow is the movement of volume per unit of time. Flow is affected by the pressure gradient and the resistance fluid encounters between 2 points. Vascular resistance is the opposition to flow, which is caused primarily by blood friction against vessel walls.Vascular Resistance, Flow, and Mean Arterial Pressure is chronic, PHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance may develop. Chronic increases in flowFlowBlood flows through the heart, arteries, capillaries, and veins in a closed, continuous circuit. Flow is the movement of volume per unit of time. Flow is affected by the pressure gradient and the resistance fluid encounters between 2 points. Vascular resistance is the opposition to flow, which is caused primarily by blood friction against vessel walls.Vascular Resistance, Flow, and Mean Arterial Pressure can also induce vascular changes leading to increased pulmonary vascular resistanceResistancePhysiologically, the opposition to flow of air caused by the forces of friction. As a part of pulmonary function testing, it is the ratio of driving pressure to the rate of air flow.Ventilation: Mechanics of Breathing.
LiverLiverThe liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: AnatomycirrhosisCirrhosisCirrhosis is a late stage of hepatic parenchymal necrosis and scarring (fibrosis) most commonly due to hepatitis C infection and alcoholic liver disease. Patients may present with jaundice, ascites, and hepatosplenomegaly. Cirrhosis can also cause complications such as hepatic encephalopathy, portal hypertension, portal vein thrombosis, and hepatorenal syndrome. Cirrhosis
Chronic anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types
Arteriovenous malformationsArteriovenous malformationsCongenital vascular anomalies in the brain characterized by direct communication between an artery and a vein without passing through the capillaries. The locations and size of the shunts determine the symptoms including headaches; seizures; stroke; intracranial hemorrhages; mass effect; and vascular steal effect.Intracerebral Hemorrhage
Familial PAHPAHThe glycine amide of 4-aminobenzoic acid. Its sodium salt is used as a diagnostic aid to measure effective renal plasma flow (ERPF) and excretory capacity.Glomerular Filtration is most commonly due to mutations in the BMPR2geneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics.
BMPR2
80% of familial cases are due to an inactivating mutationMutationGenetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations in BMPR2.
With BMPR2 inactivated, patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship are unable to prevent vascular smooth muscle proliferation → PAHPAHThe glycine amide of 4-aminobenzoic acid. Its sodium salt is used as a diagnostic aid to measure effective renal plasma flow (ERPF) and excretory capacity.Glomerular Filtration
Several other rare mutations have been identified.
Pathogenesis by classification group
Table: Pathogenesis of PHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance by classification group
FibrosisFibrosisAny pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury.Bronchiolitis Obliterans
Pulmonary vascular remodelingVascular remodelingThe active alterations of vascular wall structures, often leading to elevated vascular resistance. It is associated with aging; atherosclerosis; diabetes mellitus; hypertension; pregnancy; pulmonary hypertension; and stroke, but is also a normal part of embryogenesis.Cor Pulmonale
Reduced complianceComplianceDistensibility measure of a chamber such as the lungs (lung compliance) or bladder. Compliance is expressed as a change in volume per unit change in pressure.Veins: Histology of the pulmonary vasculature
Impaired voltage-gated potassiumPotassiumAn element in the alkali group of metals with an atomic symbol k, atomic number 19, and atomic weight 39. 10. It is the chief cation in the intracellular fluid of muscle and other cells. Potassium ion is a strong electrolyte that plays a significant role in the regulation of fluid volume and maintenance of the water-electrolyte balance.HyperkalemiachannelsChannelsThe Cell: Cell Membrane → contraction of pulmonary smooth musclesSmooth musclesUnstriated and unstriped muscle, one of the muscles of the internal organs, blood vessels, hair follicles, etc. Contractile elements are elongated, usually spindle-shaped cells with centrally located nuclei. Smooth muscle fibers are bound together into sheets or bundles by reticular fibers and frequently elastic nets are also abundant.Muscle Tissue: Histology
↑ Activity of phospholipase A2Phospholipase A2Phospholipases that hydrolyze the Acyl group attached to the 2-position of phosphoglycerides.Nephrotic Syndrome → increase in vasoconstrictive substances: vasoconstrictive prostaglandinsProstaglandinsA group of compounds derived from unsaturated 20-carbon fatty acids, primarily arachidonic acid, via the cyclooxygenase pathway. They are extremely potent mediators of a diverse group of physiological processes.Eicosanoids, thromboxanesThromboxanesPhysiologically active compounds found in many organs of the body. They are formed in vivo from the prostaglandin endoperoxides and cause platelet aggregation, contraction of arteries, and other biological effects. Thromboxanes are important mediators of the actions of polyunsaturated fatty acids transformed by cyclooxygenase.Eicosanoids, leukotrienesLeukotrienesA family of biologically active compounds derived from arachidonic acid by oxidative metabolism through the 5-lipoxygenase pathway. They participate in host defense reactions and pathophysiological conditions such as immediate hypersensitivity and inflammation. They have potent actions on many essential organs and systems, including the cardiovascular, pulmonary, and central nervous system as well as the gastrointestinal tract and the immune system.Eicosanoids
↑ Endothelin (a vasoconstrictor)
Vascular destruction due to progressive parenchymal fibrosisFibrosisAny pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury.Bronchiolitis Obliterans
Vascular inflammationInflammationInflammation is a complex set of responses to infection and injury involving leukocytes as the principal cellular mediators in the body’s defense against pathogenic organisms. Inflammation is also seen as a response to tissue injury in the process of wound healing. The 5 cardinal signs of inflammation are pain, heat, redness, swelling, and loss of function. Inflammation
Group 4: due to chronic thromboemboli
Similar to group 3
Clinical Presentation
History
Symptoms:
DyspneaDyspneaDyspnea is the subjective sensation of breathing discomfort. Dyspnea is a normal manifestation of heavy physical or psychological exertion, but also may be caused by underlying conditions (both pulmonary and extrapulmonary). Dyspnea on exertion leading to dyspneaDyspneaDyspnea is the subjective sensation of breathing discomfort. Dyspnea is a normal manifestation of heavy physical or psychological exertion, but also may be caused by underlying conditions (both pulmonary and extrapulmonary). Dyspnea at rest (primary symptom)
FatigueFatigueThe state of weariness following a period of exertion, mental or physical, characterized by a decreased capacity for work and reduced efficiency to respond to stimuli.Fibromyalgia
Chest painPainAn unpleasant sensation induced by noxious stimuli which are detected by nerve endings of nociceptive neurons.Pain: Types and Pathways (angina)
Exertional syncopeSyncopeSyncope is a short-term loss of consciousness and loss of postural stability followed by spontaneous return of consciousness to the previous neurologic baseline without the need for resuscitation. The condition is caused by transient interruption of cerebral blood flow that may be benign or related to a underlying life-threatening condition. Syncope
COPDCOPDChronic obstructive pulmonary disease (COPD) is a lung disease characterized by progressive, largely irreversible airflow obstruction. The condition usually presents in middle-aged or elderly persons with a history of cigarette smoking. Signs and symptoms include prolonged expiration, wheezing, diminished breath sounds, progressive dyspnea, and chronic cough. Chronic Obstructive Pulmonary Disease (COPD)/emphysemaEmphysemaEnlargement of air spaces distal to the terminal bronchioles where gas-exchange normally takes place. This is usually due to destruction of the alveolar wall. Pulmonary emphysema can be classified by the location and distribution of the lesions.Chronic Obstructive Pulmonary Disease (COPD)
Interstitial lung disease
Heart disease
Sickle cell anemiaSickle cell anemiaA disease characterized by chronic hemolytic anemia, episodic painful crises, and pathologic involvement of many organs. It is the clinical expression of homozygosity for hemoglobin S.Sickle Cell Disease
Travel to regions with endemic schistosomiasisSchistosomiasisInfection with flukes (trematodes) of the genus schistosoma. Three species produce the most frequent clinical diseases: Schistosoma haematobium (endemic in Africa and the Middle East), Schistosoma Mansoni (in Egypt, northern and southern Africa, some West Indies islands, northern 2/3 of South america), and Schistosoma japonicum (in Japan, China, the Philippines, Celebes, Thailand, Laos). S. mansoni is often seen in Puerto Ricans living in the United States.Schistosoma/Schistosomiasis
HypercoagulableHypercoagulableHypercoagulable states (also referred to as thrombophilias) are a group of hematologic diseases defined by an increased risk of clot formation (i.e., thrombosis) due to either an increase in procoagulants, a decrease in anticoagulants, or a decrease in fibrinolysis. Hypercoagulable States states/history of thromboembolic disease
Physical exam
Exam findings consistent with PHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance include:
Raised jugular venous pulsation (JVP)
Heart soundsHeart soundsHeart sounds are brief, transient sounds produced by valve opening and closure and by movement of blood in the heart. They are divided into systolic and diastolic sounds. In most cases, only the first (S1) and second (S2) heart sounds are heard. These are high-frequency sounds and arise from aortic and pulmonary valve closure (S1), as well as mitral and tricuspid valve closure (S2).Heart Sounds:
Pronounced 2nd heart sound (due to a louder P2 component)
Presence of extra right-sided heart soundsHeart soundsHeart sounds are brief, transient sounds produced by valve opening and closure and by movement of blood in the heart. They are divided into systolic and diastolic sounds. In most cases, only the first (S1) and second (S2) heart sounds are heard. These are high-frequency sounds and arise from aortic and pulmonary valve closure (S1), as well as mitral and tricuspid valve closure (S2).Heart Sounds:
S3S3Heart Sounds: can be heard in ventricular volume overload and heart failureHeart FailureA heterogeneous condition in which the heart is unable to pump out sufficient blood to meet the metabolic need of the body. Heart failure can be caused by structural defects, functional abnormalities (ventricular dysfunction), or a sudden overload beyond its capacity. Chronic heart failure is more common than acute heart failure which results from sudden insult to cardiac function, such as myocardial infarction.Total Anomalous Pulmonary Venous Return (TAPVR) (HF)
S4S4Heart Sounds: ↑ resistanceResistancePhysiologically, the opposition to flow of air caused by the forces of friction. As a part of pulmonary function testing, it is the ratio of driving pressure to the rate of air flow.Ventilation: Mechanics of Breathing to ventricular fillingVentricular fillingCardiac Cycle due to ↓ ventricular complianceComplianceDistensibility measure of a chamber such as the lungs (lung compliance) or bladder. Compliance is expressed as a change in volume per unit change in pressure.Veins: Histology
Peripheral edemaPeripheral edemaPeripheral edema is the swelling of the lower extremities, namely, legs, feet, and ankles.Edema
AscitesAscitesAscites is the pathologic accumulation of fluid within the peritoneal cavity that occurs due to an osmotic and/or hydrostatic pressure imbalance secondary to portal hypertension (cirrhosis, heart failure) or non-portal hypertension (hypoalbuminemia, malignancy, infection).Ascites
Pleural effusionPleural EffusionPleural effusion refers to the accumulation of fluid between the layers of the parietal and visceral pleura. Common causes of this condition include infection, malignancy, autoimmune disorders, or volume overload. Clinical manifestations include chest pain, cough, and dyspnea. Pleural Effusion
Hepatomegaly
Diagnosis
Diagnosis of group 1 PAHPAHThe glycine amide of 4-aminobenzoic acid. Its sodium salt is used as a diagnostic aid to measure effective renal plasma flow (ERPF) and excretory capacity.Glomerular Filtration is typically one of exclusion, after ruling out etiologies in groups 2–5, which can be made by the following tests:
EchocardiographyEchocardiographyUltrasonic recording of the size, motion, and composition of the heart and surrounding tissues. The standard approach is transthoracic.Tricuspid Valve Atresia (TVA)
ECGECGAn electrocardiogram (ECG) is a graphic representation of the electrical activity of the heart plotted against time. Adhesive electrodes are affixed to the skin surface allowing measurement of cardiac impulses from many angles. The ECG provides 3-dimensional information about the conduction system of the heart, the myocardium, and other cardiac structures. Electrocardiogram (ECG):
Important for:
Ruling out PHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance due to left-sided heart disease
Assessing right-sided heart function
Findings of right heart disease may include:
TachycardiaTachycardiaAbnormally rapid heartbeat, usually with a heart rate above 100 beats per minute for adults. Tachycardia accompanied by disturbance in the cardiac depolarization (cardiac arrhythmia) is called tachyarrhythmia.Sepsis in Children
Right axis deviation
Upright R waves in V1–V3
Chest X-rayX-rayPenetrating electromagnetic radiation emitted when the inner orbital electrons of an atom are excited and release radiant energy. X-ray wavelengths range from 1 pm to 10 nm. Hard x-rays are the higher energy, shorter wavelength x-rays. Soft x-rays or grenz rays are less energetic and longer in wavelength. The short wavelength end of the x-ray spectrum overlaps the gamma rays wavelength range. The distinction between gamma rays and x-rays is based on their radiation source.Pulmonary Function Tests findings:
Enlargement of central pulmonary arteriesArteriesArteries are tubular collections of cells that transport oxygenated blood and nutrients from the heart to the tissues of the body. The blood passes through the arteries in order of decreasing luminal diameter, starting in the largest artery (the aorta) and ending in the small arterioles. Arteries are classified into 3 types: large elastic arteries, medium muscular arteries, and small arteries and arterioles. Arteries: Histology and the main branches
Tapering of distal vessels
Findings consistent with heart disease: cardiomegalyCardiomegalyEnlargement of the heart, usually indicated by a cardiothoracic ratio above 0. 50. Heart enlargement may involve the right, the left, or both heart ventricles or heart atria. Cardiomegaly is a nonspecific symptom seen in patients with chronic systolic heart failure (heart failure) or several forms of cardiomyopathies.Ebstein’s Anomaly, pulmonary edemaPulmonary edemaPulmonary edema is a condition caused by excess fluid within the lung parenchyma and alveoli as a consequence of a disease process. Based on etiology, pulmonary edema is classified as cardiogenic or noncardiogenic. Patients may present with progressive dyspnea, orthopnea, cough, or respiratory failure.Pulmonary Edema
Findings consistent with lung disease: COPDCOPDChronic obstructive pulmonary disease (COPD) is a lung disease characterized by progressive, largely irreversible airflow obstruction. The condition usually presents in middle-aged or elderly persons with a history of cigarette smoking. Signs and symptoms include prolonged expiration, wheezing, diminished breath sounds, progressive dyspnea, and chronic cough. Chronic Obstructive Pulmonary Disease (COPD), interstitial lung disease
Note: Chest X-rayX-rayPenetrating electromagnetic radiation emitted when the inner orbital electrons of an atom are excited and release radiant energy. X-ray wavelengths range from 1 pm to 10 nm. Hard x-rays are the higher energy, shorter wavelength x-rays. Soft x-rays or grenz rays are less energetic and longer in wavelength. The short wavelength end of the x-ray spectrum overlaps the gamma rays wavelength range. The distinction between gamma rays and x-rays is based on their radiation source.Pulmonary Function Tests is often normal or with minimal findings.
Ventilation-perfusion lung scan:
Helps differentiate group 3 (lung/hypoxic disease) from group 4 (chronic thromboembolic disease)
Findings may include:
Group 3: diffuse mottled perfusion
Group 4: segmental mismatched defects
Pulmonary function tests:
Useful in diagnosing lung and/or hypoxic disorders
Obstructive patterns: COPDCOPDChronic obstructive pulmonary disease (COPD) is a lung disease characterized by progressive, largely irreversible airflow obstruction. The condition usually presents in middle-aged or elderly persons with a history of cigarette smoking. Signs and symptoms include prolonged expiration, wheezing, diminished breath sounds, progressive dyspnea, and chronic cough. Chronic Obstructive Pulmonary Disease (COPD)
Restrictive patterns: interstitial lung disease
Right-sided cardiac catheterizationCardiac CatheterizationProcedures in which placement of cardiac catheters is performed for therapeutic or diagnostic procedures.Cardiac Surgery:
Most accurate test for confirming the diagnosis, especially in idiopathicIdiopathicDermatomyositis cases of PAHPAHThe glycine amide of 4-aminobenzoic acid. Its sodium salt is used as a diagnostic aid to measure effective renal plasma flow (ERPF) and excretory capacity.Glomerular Filtration
Cardiac outputCardiac outputThe volume of blood passing through the heart per unit of time. It is usually expressed as liters (volume) per minute so as not to be confused with stroke volume (volume per beat).Cardiac Mechanics
Cardiac shunting
Other causes of dyspneaDyspneaDyspnea is the subjective sensation of breathing discomfort. Dyspnea is a normal manifestation of heavy physical or psychological exertion, but also may be caused by underlying conditions (both pulmonary and extrapulmonary). Dyspnea or angina
Allows for testing of vasoreactivity in patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship with group 1 PAHPAHThe glycine amide of 4-aminobenzoic acid. Its sodium salt is used as a diagnostic aid to measure effective renal plasma flow (ERPF) and excretory capacity.Glomerular Filtration:
Administer a short-acting vasodilatory substance (e.g., inhaled NO) and assess response.
Important for managing patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship with group 1 PAHPAHThe glycine amide of 4-aminobenzoic acid. Its sodium salt is used as a diagnostic aid to measure effective renal plasma flow (ERPF) and excretory capacity.Glomerular Filtration
Laboratory tests: can help identify other causes of PHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance
CBC: to rule out anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types
Liver function testsLiver function testsLiver function tests, also known as hepatic function panels, are one of the most commonly performed screening blood tests. Such tests are also used to detect, evaluate, and monitor acute and chronic liver diseases.Liver Function Tests: to rule out liverLiverThe liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy disease as a cause of symptoms
ANA: test to screen for sclerodermaSclerodermaScleroderma (systemic sclerosis) is an autoimmune condition characterized by diffuse collagen deposition and fibrosis. The clinical presentation varies from limited skin involvement to diffuse involvement of internal organs. Scleroderma
Assays for schistosomiasisSchistosomiasisInfection with flukes (trematodes) of the genus schistosoma. Three species produce the most frequent clinical diseases: Schistosoma haematobium (endemic in Africa and the Middle East), Schistosoma Mansoni (in Egypt, northern and southern Africa, some West Indies islands, northern 2/3 of South america), and Schistosoma japonicum (in Japan, China, the Philippines, Celebes, Thailand, Laos). S. mansoni is often seen in Puerto Ricans living in the United States.Schistosoma/Schistosomiasis
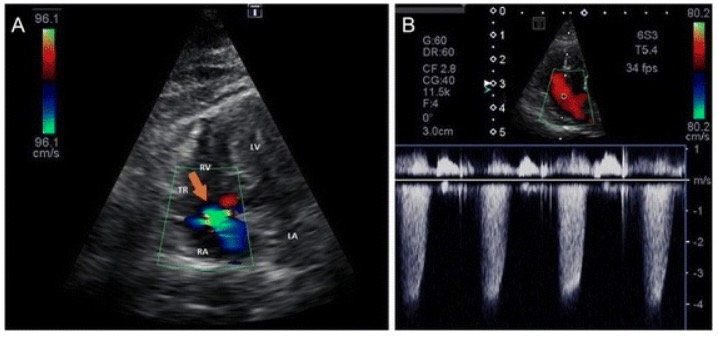
Tricuspid regurgitation: Tricuspid regurgitation approximates 4.2 m/sec, indicating a peak tricuspid regurgitation pressure gradient of approximately 70 mm Hg (moderate pulmonary hypertension) A: Color Doppler image of tricuspid regurgitation B: Continuous Doppler (CW) image from the left apical 4-chamber view optimized for the right ventricle
LA: left atrium LV: left ventricle RA: right atrium RV: right ventricle TR: tricuspid regurgitation (orange arrow)
Image: “Tricuspid regurgitation” by J.A. Jaffey et al. License: CC BY 4.0
For groups 2–5, management should be directed at treating the underlying condition. In addition, management should focus on maintaining/improving oxygenation. PatientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship should be referred to specialists at tertiary centers for management, which is often complex.
Vasodilatory agents
CalciumCalciumA basic element found in nearly all tissues. It is a member of the alkaline earth family of metals with the atomic symbol ca, atomic number 20, and atomic weight 40. Calcium is the most abundant mineral in the body and combines with phosphorus to form calcium phosphate in the bones and teeth. It is essential for the normal functioning of nerves and muscles and plays a role in blood coagulation (as factor IV) and in many enzymatic processes.Electrolytes channel blockers (CCBsCCBsCalcium channel blockers (CCBS) are a class of medications that inhibit voltage-dependent L-type calcium channels of cardiac and vascular smooth muscle cells. The inhibition of these channels produces vasodilation and myocardial depression. There are 2 major classes of CCBS: dihydropyridines and non-dihydropyridines.Class 4 Antiarrhythmic Drugs (Calcium Channel Blockers)):
Only effective in patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship who are vasoreactive
CCBsCCBsCalcium channel blockers (CCBS) are a class of medications that inhibit voltage-dependent L-type calcium channels of cardiac and vascular smooth muscle cells. The inhibition of these channels produces vasodilation and myocardial depression. There are 2 major classes of CCBS: dihydropyridines and non-dihydropyridines.Class 4 Antiarrhythmic Drugs (Calcium Channel Blockers) block the inward movement of calciumCalciumA basic element found in nearly all tissues. It is a member of the alkaline earth family of metals with the atomic symbol ca, atomic number 20, and atomic weight 40. Calcium is the most abundant mineral in the body and combines with phosphorus to form calcium phosphate in the bones and teeth. It is essential for the normal functioning of nerves and muscles and plays a role in blood coagulation (as factor IV) and in many enzymatic processes.Electrolytes into:
Endothelial cells → vasodilationVasodilationThe physiological widening of blood vessels by relaxing the underlying vascular smooth muscle.Pulmonary Hypertension Drugs
Sinoatrial (SA) and atrioventricular (AV) nodes → ↓ cardiac conduction and contractility
Examples: amlodipineAmlodipineA long-acting dihydropyridine calcium channel blocker. It is effective in the treatment of angina pectoris and hypertension.Hypertension Drugs, nifedipineNifedipineA potent vasodilator agent with calcium antagonistic action. It is a useful anti-anginal agent that also lowers blood pressure.Class 4 Antiarrhythmic Drugs (Calcium Channel Blockers)
High doses may produce a dramatic reduction in pulmonary arteryPulmonary arteryThe short wide vessel arising from the conus arteriosus of the right ventricle and conveying unaerated blood to the lungs.Lungs: Anatomy pressure.
Vasodilatory prostaglandinsProstaglandinsA group of compounds derived from unsaturated 20-carbon fatty acids, primarily arachidonic acid, via the cyclooxygenase pathway. They are extremely potent mediators of a diverse group of physiological processes.Eicosanoids:
ProstacyclinProstacyclinA prostaglandin that is a powerful vasodilator and inhibits platelet aggregation. It is biosynthesized enzymatically from prostaglandin endoperoxides in human vascular tissue. The sodium salt has been also used to treat primary pulmonary hypertension.Eicosanoids (PGIPGIAn aldose-ketose isomerase that catalyzes the reversible interconversion of glucose 6-phosphate and fructose 6-phosphate. In prokaryotic and eukaryotic organisms it plays an essential role in glycolytic and gluconeogenic pathways. In mammalian systems the enzyme is found in the cytoplasm and as a secreted protein. This secreted form of glucose-6-phosphate isomerase has been referred to as autocrine motility factor or neuroleukin, and acts as a cytokine which binds to the autocrine motility factor receptor. Deficiency of the enzyme in humans is an autosomal recessive trait, which results in congenital nonspherocytic hemolytic anemia.Glycolysis2) is a natural vasodilatory substance.
PGIPGIAn aldose-ketose isomerase that catalyzes the reversible interconversion of glucose 6-phosphate and fructose 6-phosphate. In prokaryotic and eukaryotic organisms it plays an essential role in glycolytic and gluconeogenic pathways. In mammalian systems the enzyme is found in the cytoplasm and as a secreted protein. This secreted form of glucose-6-phosphate isomerase has been referred to as autocrine motility factor or neuroleukin, and acts as a cytokine which binds to the autocrine motility factor receptor. Deficiency of the enzyme in humans is an autosomal recessive trait, which results in congenital nonspherocytic hemolytic anemia.Glycolysis2 analogs: epoprostenolEpoprostenolA prostaglandin that is a powerful vasodilator and inhibits platelet aggregation. It is biosynthesized enzymatically from prostaglandin endoperoxides in human vascular tissue. The sodium salt has been also used to treat primary pulmonary hypertension.Hemostasis, treprostinilTreprostinilPulmonary Hypertension Drugs
EndothelinsEndothelins21-amino-acid peptides produced by vascular endothelial cells and functioning as potent vasoconstrictors. The endothelin family consists of three members, endothelin-1; endothelin-2; and endothelin-3. All three peptides contain 21 amino acids, but vary in amino acid composition. The three peptides produce vasoconstrictor and pressor responses in various parts of the body. However, the quantitative profiles of the pharmacological activities are considerably different among the three isopeptides.Hemostasis are natural vasoconstrictors.
Competitively antagonize endothelin receptorsReceptorsReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors → ↓ pulmonary vascular resistanceResistancePhysiologically, the opposition to flow of air caused by the forces of friction. As a part of pulmonary function testing, it is the ratio of driving pressure to the rate of air flow.Ventilation: Mechanics of Breathing
Phosphodiesterase inhibitorsPhosphodiesterase inhibitorsPhosphodiesterase (PDE) inhibitors are a group of drugs that act by inhibiting PDE enzymes. Phosphodiesterase inhibitors have various mechanisms of action depending on the subtype of PDE targeted, but their main action is increasing the amount of intracellular cAMP or cGMP, which in turn results in physiologic effects such as reducing inflammation, promoting smooth muscle relaxation, and vasodilation.Phosphodiesterase Inhibitors (PDE-5 inhibitorsPDE-5 inhibitorsPulmonary Hypertension Drugs):
Results in ↑ pulmonary and systemic vasodilationVasodilationThe physiological widening of blood vessels by relaxing the underlying vascular smooth muscle.Pulmonary Hypertension Drugs
Examples: sildenafilSildenafilA phosphodiesterase type-5 inhibitor; vasodilator agent and urological agent that is used in the treatment of erectile dysfunction and primary pulmonary hypertension.Phosphodiesterase Inhibitors (Viagra), tadalafilTadalafilA carboline derivative and phosphodiesterase 5 inhibitor that is used primarily to treat erectile dysfunction; benign prostatic hyperplasia and primary pulmonary hypertension.Phosphodiesterase Inhibitors
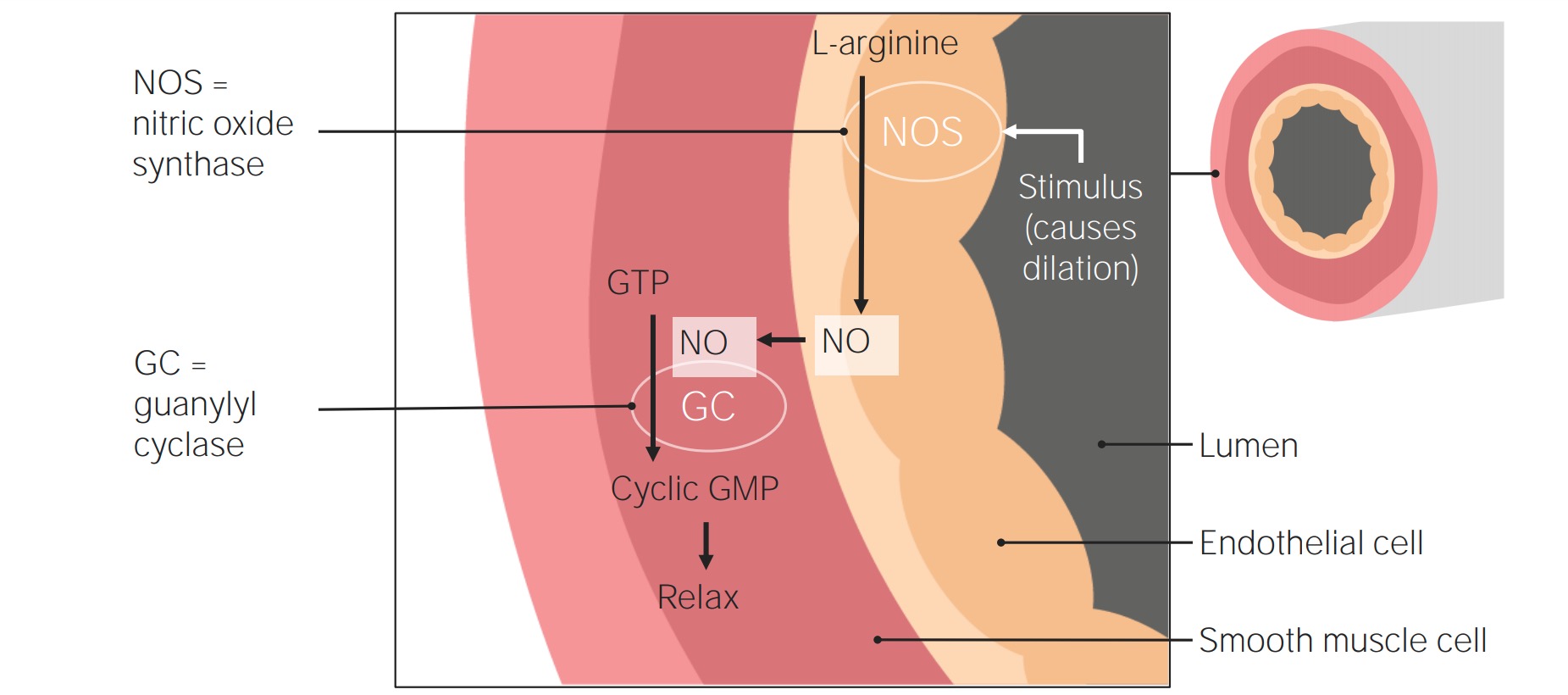
Soluble guanylate cyclaseGuanylate cyclaseAn enzyme that catalyzes the conversion of GTP to 3.Diarrheagenic E. coli (sGC) is an intracellular receptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors for NO.
Vasodilatory agents for the treatment of pulmonary hypertension: A stimulus triggers NO synthase (NOS) in the endothelial cells to convert L-arginine into NO. The NO then moves into the smooth muscle, where it stimulates the activity of guanylate cyclase (also known as guanylyl cyclase), which converts guanosine triphosphate into cGMP. Cyclic GMP then induces smooth muscle relaxation, resulting in vasodilation.
InfluenzaInfluenzaInfluenza viruses are members of the Orthomyxoviridae family and the causative organisms of influenza, a highly contagious febrile respiratory disease. There are 3 primary influenza viruses (A, B, and C) and various subtypes, which are classified based on their virulent surface antigens, hemagglutinin (HA) and neuraminidase (NA). Influenza typically presents with a fever, myalgia, headache, and symptoms of an upper respiratory infection. Influenza Viruses/InfluenzavaccinationVaccinationVaccination is the administration of a substance to induce the immune system to develop protection against a disease. Unlike passive immunization, which involves the administration of pre-performed antibodies, active immunization constitutes the administration of a vaccine to stimulate the body to produce its own antibodies.Vaccination
Birth control to prevent pregnancyPregnancyThe status during which female mammals carry their developing young (embryos or fetuses) in utero before birth, beginning from fertilization to birth.Pregnancy: Diagnosis, Physiology, and Care due to high risk of maternal mortalityMortalityAll deaths reported in a given population.Measures of Health Status associated with PHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance in pregnancyPregnancyThe status during which female mammals carry their developing young (embryos or fetuses) in utero before birth, beginning from fertilization to birth.Pregnancy: Diagnosis, Physiology, and Care
Indication: patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship with right-sided volume overload (edemaEdemaEdema is a condition in which excess serous fluid accumulates in the body cavity or interstitial space of connective tissues. Edema is a symptom observed in several medical conditions. It can be categorized into 2 types, namely, peripheral (in the extremities) and internal (in an organ or body cavity). Edema, ascitesAscitesAscites is the pathologic accumulation of fluid within the peritoneal cavity that occurs due to an osmotic and/or hydrostatic pressure imbalance secondary to portal hypertension (cirrhosis, heart failure) or non-portal hypertension (hypoalbuminemia, malignancy, infection).Ascites)
Loop diureticsDiureticsAgents that promote the excretion of urine through their effects on kidney function.Heart Failure and Chronic Coronary Syndrome Medication are typically 1st-line drugs (e.g., furosemideFurosemideA benzoic-sulfonamide-furan. It is a diuretic with fast onset and short duration that is used for edema and chronic renal insufficiency.Loop Diuretics).
Group 4 (PHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance due to chronic thromboembolic disease)
Group 1 PAHPAHThe glycine amide of 4-aminobenzoic acid. Its sodium salt is used as a diagnostic aid to measure effective renal plasma flow (ERPF) and excretory capacity.Glomerular Filtration that is either idiopathicIdiopathicDermatomyositis or familial
Atrial fibrillationAtrial fibrillationAtrial fibrillation (AF or Afib) is a supraventricular tachyarrhythmia and the most common kind of arrhythmia. It is caused by rapid, uncontrolled atrial contractions and uncoordinated ventricular responses. Atrial Fibrillation
WarfarinWarfarinAn anticoagulant that acts by inhibiting the synthesis of vitamin K-dependent coagulation factors. Warfarin is indicated for the prophylaxis and/or treatment of venous thrombosis and its extension, pulmonary embolism, and atrial fibrillation with embolization. It is also used as an adjunct in the prophylaxis of systemic embolism after myocardial infarction. Warfarin is also used as a rodenticide.Anticoagulants is usually the drug of choice.
DigoxinDigoxinA cardiotonic glycoside obtained mainly from digitalis lanata; it consists of three sugars and the aglycone digoxigenin. Digoxin has positive inotropic and negative chronotropic activity. It is used to control ventricular rate in atrial fibrillation and in the management of congestive heart failure with atrial fibrillation. Its use in congestive heart failure and sinus rhythm is less certain. The margin between toxic and therapeutic doses is small.Cardiac Glycosides:
Indications:
Heart failureHeart FailureA heterogeneous condition in which the heart is unable to pump out sufficient blood to meet the metabolic need of the body. Heart failure can be caused by structural defects, functional abnormalities (ventricular dysfunction), or a sudden overload beyond its capacity. Chronic heart failure is more common than acute heart failure which results from sudden insult to cardiac function, such as myocardial infarction.Total Anomalous Pulmonary Venous Return (TAPVR) due to systolic dysfunctionSystolic dysfunctionDilated Cardiomyopathy
Use of parenteral prostaglandinsProstaglandinsA group of compounds derived from unsaturated 20-carbon fatty acids, primarily arachidonic acid, via the cyclooxygenase pathway. They are extremely potent mediators of a diverse group of physiological processes.Eicosanoids
Known or suspected pulmonary venoocclusive disease
Procedures of choice:
Bilateral lung transplant
Heart-lung transplant
Creation of a right-to-left shunt
Can be temporizing (as a bridge to transplant) or palliative
Endovascular thrombectomyThrombectomySurgical removal of an obstructing clot or foreign material from a blood vessel at the point of its formation. Removal of a clot arising from a distant site is called embolectomy.Vascular Surgery: in patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship with chronic thromboembolismThromboembolismObstruction of a blood vessel (embolism) by a blood clot (thrombus) in the blood stream.Systemic Lupus Erythematosus and a known source
PrognosisPrognosisA prediction of the probable outcome of a disease based on a individual’s condition and the usual course of the disease as seen in similar situations.Non-Hodgkin Lymphomas
PAHPAHThe glycine amide of 4-aminobenzoic acid. Its sodium salt is used as a diagnostic aid to measure effective renal plasma flow (ERPF) and excretory capacity.Glomerular Filtration is progressive and may be fatal if left untreated.
Untreated, group 1 PAHPAHThe glycine amide of 4-aminobenzoic acid. Its sodium salt is used as a diagnostic aid to measure effective renal plasma flow (ERPF) and excretory capacity.Glomerular Filtration has the worst prognosisPrognosisA prediction of the probable outcome of a disease based on a individual’s condition and the usual course of the disease as seen in similar situations.Non-Hodgkin Lymphomas:
1-year survival: 85%
3-year survival: 68%
5-year survival: 57%
PrognosisPrognosisA prediction of the probable outcome of a disease based on a individual’s condition and the usual course of the disease as seen in similar situations.Non-Hodgkin Lymphomas depends on the underlying cause.
The main cause of death is right ventricular failure.
PatientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship should be monitored by a specialist to assess for a decline in functional status.
HF: the inability of the heart to supply the body with normal cardiac outputCardiac outputThe volume of blood passing through the heart per unit of time. It is usually expressed as liters (volume) per minute so as not to be confused with stroke volume (volume per beat).Cardiac Mechanics to meet metabolic needs. Heart failureHeart FailureA heterogeneous condition in which the heart is unable to pump out sufficient blood to meet the metabolic need of the body. Heart failure can be caused by structural defects, functional abnormalities (ventricular dysfunction), or a sudden overload beyond its capacity. Chronic heart failure is more common than acute heart failure which results from sudden insult to cardiac function, such as myocardial infarction.Total Anomalous Pulmonary Venous Return (TAPVR) can lead to group 2 PHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance. EchocardiographyEchocardiographyUltrasonic recording of the size, motion, and composition of the heart and surrounding tissues. The standard approach is transthoracic.Tricuspid Valve Atresia (TVA) can confirm the diagnosis and provide information about the EFEFCardiac Cycle. Treatment is directed at the removal of excess fluid and decreasing oxygen demand of the heart.
Coronary arteryCoronary ArteryTruncus Arteriosus disease: occurs due to a stenosisStenosisHypoplastic Left Heart Syndrome (HLHS) of the coronary arteriesArteriesArteries are tubular collections of cells that transport oxygenated blood and nutrients from the heart to the tissues of the body. The blood passes through the arteries in order of decreasing luminal diameter, starting in the largest artery (the aorta) and ending in the small arterioles. Arteries are classified into 3 types: large elastic arteries, medium muscular arteries, and small arteries and arterioles. Arteries: Histology, leading to ischemiaIschemiaA hypoperfusion of the blood through an organ or tissue caused by a pathologic constriction or obstruction of its blood vessels, or an absence of blood circulation.Ischemic Cell Damage of the heart. Symptoms include chest painPainAn unpleasant sensation induced by noxious stimuli which are detected by nerve endings of nociceptive neurons.Pain: Types and Pathways and dyspneaDyspneaDyspnea is the subjective sensation of breathing discomfort. Dyspnea is a normal manifestation of heavy physical or psychological exertion, but also may be caused by underlying conditions (both pulmonary and extrapulmonary). Dyspnea. Diagnosis is based on history, ECGECGAn electrocardiogram (ECG) is a graphic representation of the electrical activity of the heart plotted against time. Adhesive electrodes are affixed to the skin surface allowing measurement of cardiac impulses from many angles. The ECG provides 3-dimensional information about the conduction system of the heart, the myocardium, and other cardiac structures. Electrocardiogram (ECG) findings, cardiac stress tests, and/or cardiac catheterizations. Treatment is primarily based on reducing oxygen demand of the heart and increasing the delivery of oxygen.
Tricuspid regurgitationRegurgitationGastroesophageal Reflux Disease (GERD) (TR): a valvular defect that allows the backflow of blood from the right ventricle to the right atrium during systoleSystolePeriod of contraction of the heart, especially of the heart ventricles.Cardiac Cycle.Tricuspid regurgitationRegurgitationGastroesophageal Reflux Disease (GERD) may be asymptomatic or present with systemic venous congestion due to increased right atrial and venous pressures. Tricuspid regurgitationRegurgitationGastroesophageal Reflux Disease (GERD) can also result from PHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance, but intrinsic valve disease should be considered in the differential diagnosis of PHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance. EchocardiographyEchocardiographyUltrasonic recording of the size, motion, and composition of the heart and surrounding tissues. The standard approach is transthoracic.Tricuspid Valve Atresia (TVA) can help establish the diagnosis. Treatment focuses on heart failureHeart FailureA heterogeneous condition in which the heart is unable to pump out sufficient blood to meet the metabolic need of the body. Heart failure can be caused by structural defects, functional abnormalities (ventricular dysfunction), or a sudden overload beyond its capacity. Chronic heart failure is more common than acute heart failure which results from sudden insult to cardiac function, such as myocardial infarction.Total Anomalous Pulmonary Venous Return (TAPVR) management, and surgery is reserved for severe disease.
Pulmonary fibrosisFibrosisAny pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury.Bronchiolitis Obliterans: a rapidly progressive interstitial lung disease with few available therapies. As pulmonary fibrosisFibrosisAny pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury.Bronchiolitis Obliterans progresses, group 3 PHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance can develop. The average life expectancyLife expectancyBased on known statistical data, the number of years which any person of a given age may reasonably expected to live.Population Pyramids is 3–4 years from diagnosis. Lung transplantationLung transplantationThe transference of either one or both of the lungs from one human or animal to another.Organ Transplantation is the only curative intervention, provided candidacy.
COPDCOPDChronic obstructive pulmonary disease (COPD) is a lung disease characterized by progressive, largely irreversible airflow obstruction. The condition usually presents in middle-aged or elderly persons with a history of cigarette smoking. Signs and symptoms include prolonged expiration, wheezing, diminished breath sounds, progressive dyspnea, and chronic cough. Chronic Obstructive Pulmonary Disease (COPD): a lung disease usually caused by smokingSmokingWillful or deliberate act of inhaling and exhaling smoke from burning substances or agents held by hand.Interstitial Lung Diseases, and characterized by progressive, largely irreversible airflow obstruction secondary to chronic inflammationChronic InflammationInflammation. Chronic obstructive pulmonary diseasePulmonary diseaseDiseases involving the respiratory system.Blastomyces/Blastomycosis is one of the primary causes of group 3 PHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance and the symptoms include progressive dyspneaDyspneaDyspnea is the subjective sensation of breathing discomfort. Dyspnea is a normal manifestation of heavy physical or psychological exertion, but also may be caused by underlying conditions (both pulmonary and extrapulmonary). Dyspnea and chronic cough. The diagnosis is confirmed with a pulmonary function testPulmonary function testPulmonary function tests are a group of diagnostic procedures yielding useful, quantifiable information about the rate of the flow of air through the individual’s airways, lung capacity, and the efficiency of gas exchange in relation to time. The most commonly utilized tests include spirometry (before and after bronchodilator use), lung volumes, and quantitation of diffusing capacity for carbon monoxide (CO). The tests can be influenced by the individual’s effort/fatigue, disease state, or anatomical malformation.Pulmonary Function Tests. Management includes smokingSmokingWillful or deliberate act of inhaling and exhaling smoke from burning substances or agents held by hand.Interstitial Lung Diseases cessation, pulmonary rehabilitation, and pharmacotherapy.
Pulmonary embolismPulmonary EmbolismPulmonary embolism (PE) is a potentially fatal condition that occurs as a result of intraluminal obstruction of the main pulmonary artery or its branches. The causative factors include thrombi, air, amniotic fluid, and fat. In PE, gas exchange is impaired due to the decreased return of deoxygenated blood to the lungs. Pulmonary Embolism: a result of the intraluminal obstruction of the main pulmonary arteryPulmonary arteryThe short wide vessel arising from the conus arteriosus of the right ventricle and conveying unaerated blood to the lungs.Lungs: Anatomy or its branches by certain components (e.g., thrombus, cholesterolCholesterolThe principal sterol of all higher animals, distributed in body tissues, especially the brain and spinal cord, and in animal fats and oils.Cholesterol Metabolism, air, amniotic fluidAmniotic fluidA clear, yellowish liquid that envelopes the fetus inside the sac of amnion. In the first trimester, it is likely a transudate of maternal or fetal plasma. In the second trimester, amniotic fluid derives primarily from fetal lung and kidney. Cells or substances in this fluid can be removed for prenatal diagnostic tests (amniocentesis).Placenta, Umbilical Cord, and Amniotic Cavity, or fat). The most common presenting symptom is dyspneaDyspneaDyspnea is the subjective sensation of breathing discomfort. Dyspnea is a normal manifestation of heavy physical or psychological exertion, but also may be caused by underlying conditions (both pulmonary and extrapulmonary). Dyspnea. Initial management is supportive (focusing on restoring oxygenation and hemodynamic stability), which is followed by systemic anticoagulationAnticoagulationPulmonary Hypertension Drugs and interventional therapies. Chronic thromboembolic events can lead to group 4 PHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance.
Simonneau, G., Gatzoulis, M.A., Adatia, I., Celermajer, D., Denton, C., Ghofrani, A., Gomez Sanchez, M.A., Krishna Kumar, R., Landzberg, M., Machado, R.F., Olschewski, H., Robbins, I.M., Souza, R. (2013). Updated clinical classification of pulmonary hypertension. Journal of the American College of Cardiology, 62(25 Suppl), D34–D41. https://doi.org/10.1016/j.jacc.2013.10.029